Порочный круг нейродегенерации
БиомолекулаЧто вызывает такие драматические последствия в мозге, и в конечном счете — его неминуемую гибель? За нейродегенерацией стоит целый ряд, а лучше сказать — спектр молекулярных патологических процессов. Обычно они встречаются не по отдельности, а «ходят вместе под руку» и усиливают разрушительный эффект друг друга, будучи связанными по принципу порочного круга (положительными обратными связями). Среди таких факторов стоит выделить:
- накопление патологических агрегатов белков,
- нарушенный протеостаз,
- изменения цитоскелета,
- патологию энергетического метаболизма,
- повреждения ДНК и РНК,
- нейровоспаление,
и, как следствие
- патологию синапсов,
- разобщение нейронных сетей,
и, last but not least,
- гибель нейронов.
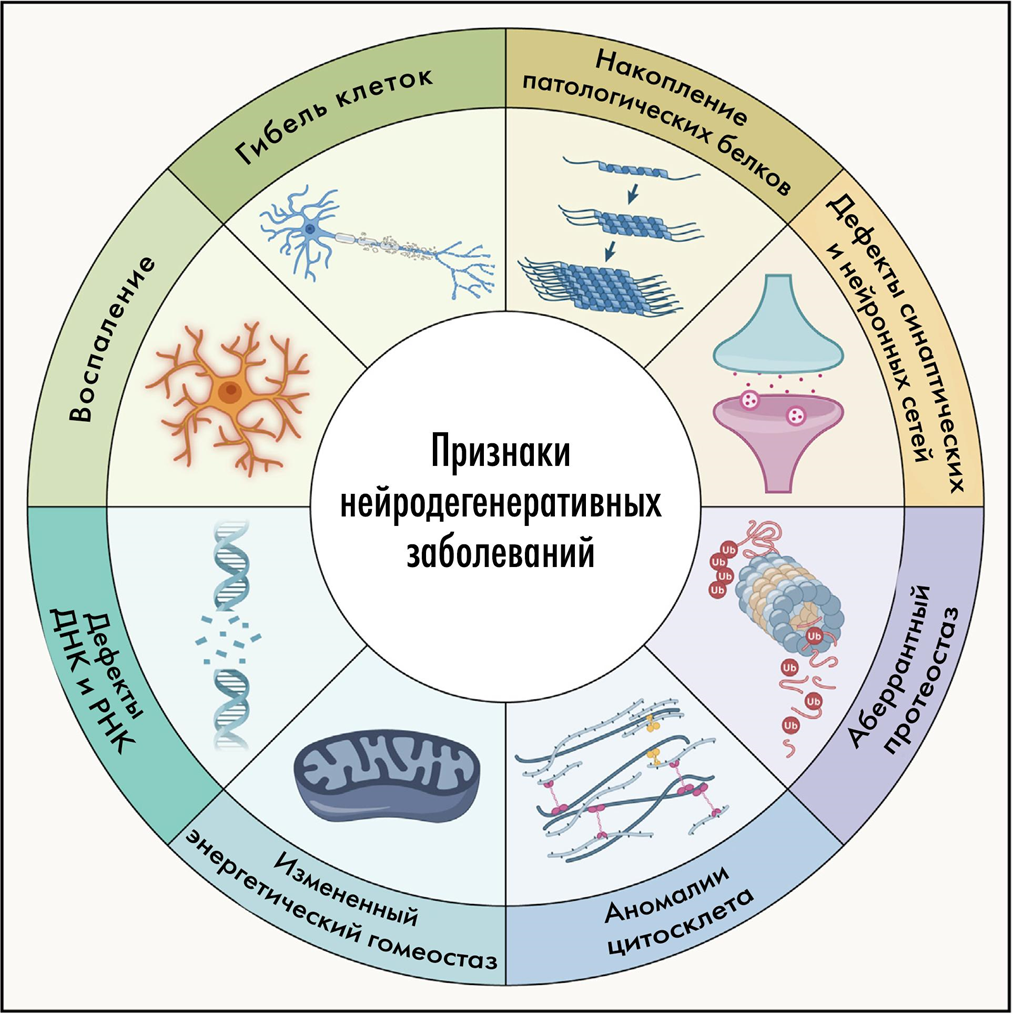
Давайте теперь пройдемся по «порочному кругу» и кратко опишем его основные компоненты.
Накопление патологических агрегатов белков
Пожалуй, самый важный и универсальный патогенетический механизм нейродегенерации — проблемы с белками. Неправильно свернутые белки могут вызывать различные заболевания и передавать это свойство соседним молекулам.
Болезни, которые вызваны таким распространением патологической конформации белка, называют конформационными, или протеинопатиями. К ним относятся многие нейродегенерации.
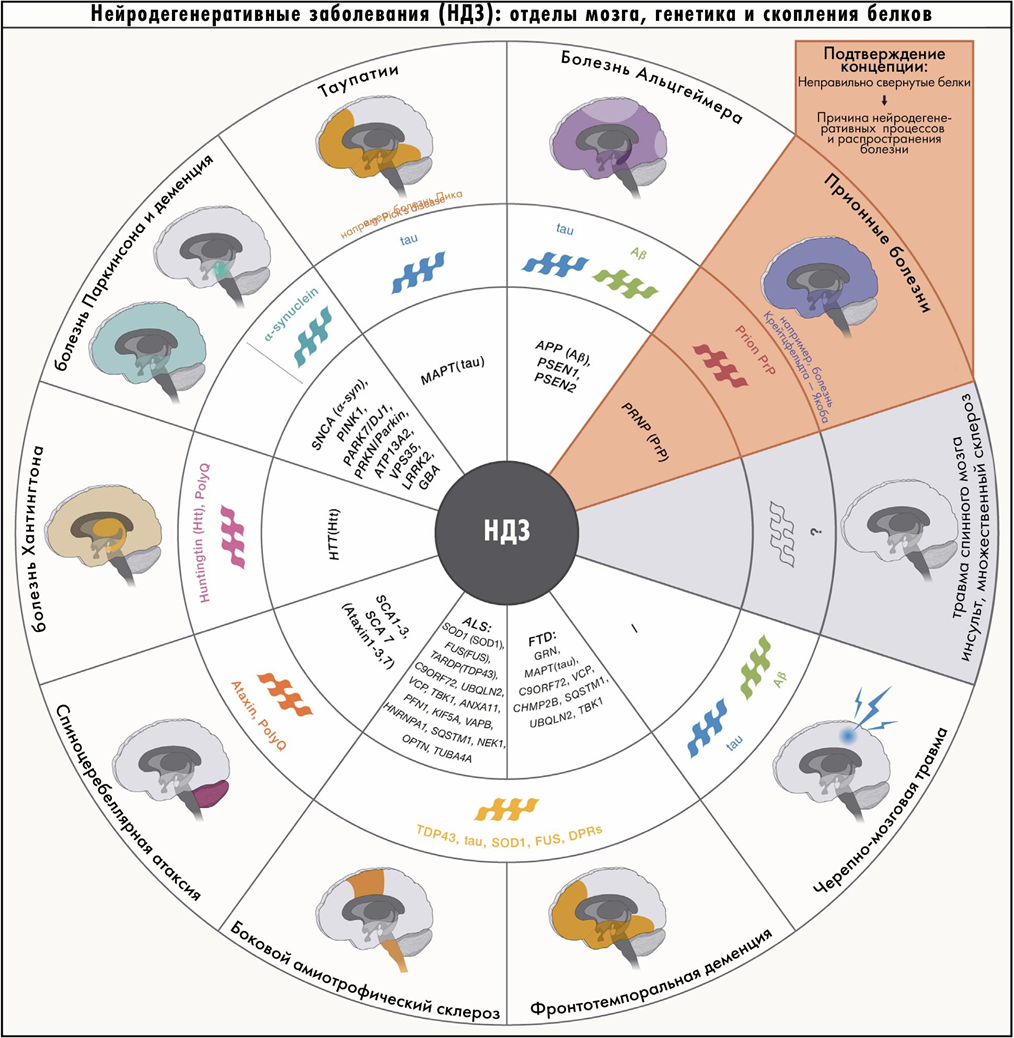
Таупатии связаны с ассоциированным с микротрубочками белком тау (ген MAPT, microtubule-associated protein tau), который собирает и стабилизирует цитоскелет нейронов. Белок имеет ряд изоформ, среди которых важнее всего две, отличающиеся числом тандемных повторов. Им соответствуют 3R-таупатии (лобно-височная деменция, в патогенезе участвует только 3R-форма), 4R-таупатии (прогрессирующий супрануклеарный паралич, кортикобазальная дегенерация и др. — только 4R-форма) и смешанные таупатии (болезнь Альцгеймера, при которой важны обе изоформы).
Вторая группа — синуклеинопатии — связаны с патологическими изменениями белков синуклеинов в мозге, главным образом альфа-синуклеина. Белок задействован в направленном транспорте веществ в везикулах и выделении нейромедиаторов в синапс. Сюда относятся болезнь Паркинсона, мультисистемная атрофия и деменция с тельцами Леви.
Нарушенный протеостаз
Различные сломанные, неправильно свернутые и не до конца синтезированные белки для клетки — дело житейское. Конечно, если их не слишком много. Такой мусор есть кому убирать — им заняты две главные системы так называемого протеостаза, то есть поддержания протеома в должном состоянии. Первая — система убиквитинилирования протеасом, которая присоединяет к «неправильным» белковым молекулам остатки молекулы-метки (убиквитина) и транспортирует к особым белковым комплексам (протеасомам), где они деградируют. Вторая — система аутофагии и лизосом, — специализируется на «переваривании» в мембранных органеллах более крупных агрегатов белков и целых органелл (например, митохондрий). Она работает в том числе в синапсах и аксонах нейронов. Обычно обе системы-«уборщики» активируются в ответ на стресс в условиях голодания, а их поломка чревата запуском механизмов клеточной гибели.
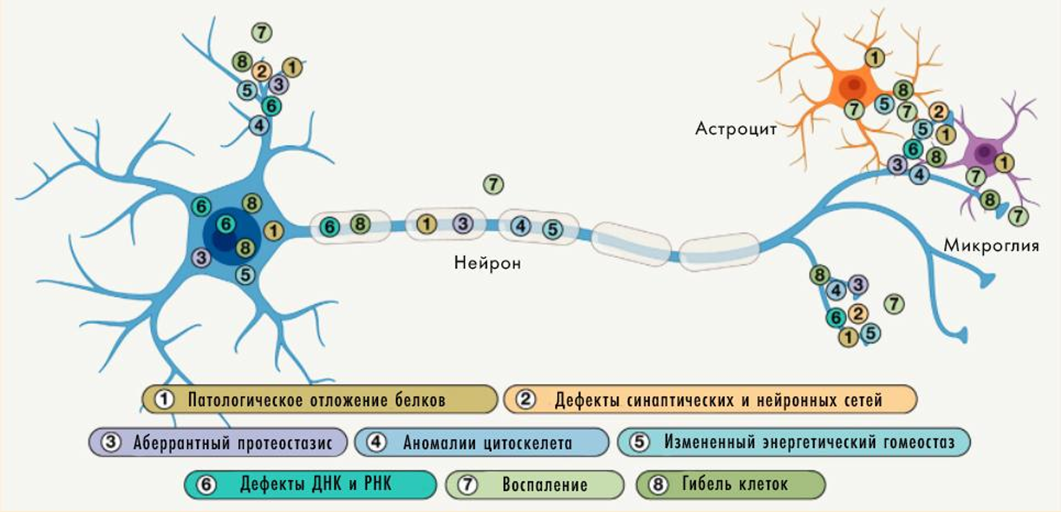
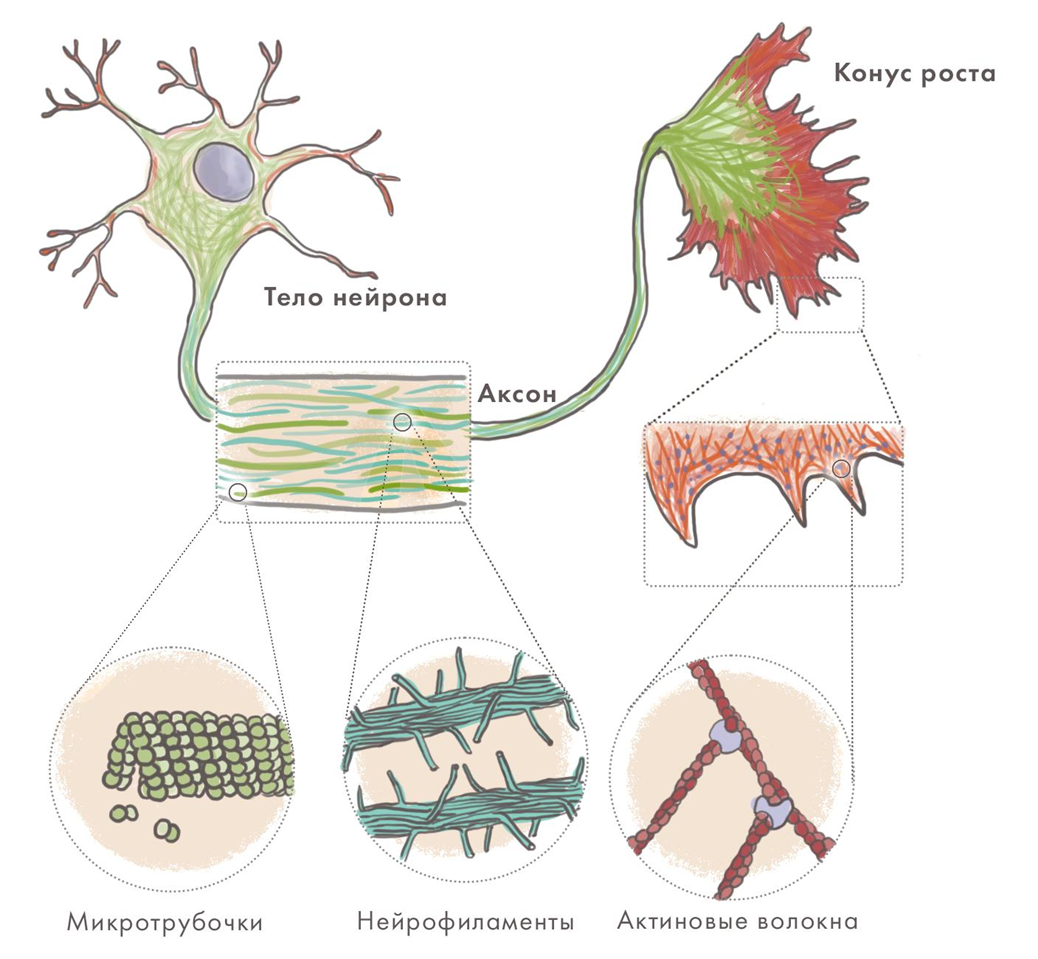
Изменения цитоскелета
Нейроны имеют сложную ультраструктуру, которую обеспечивает развитый и динамичный цитоскелет. Он образован структурами трех типов: микротрубочками, промежуточными филаментами и микрофиламентами из актина. Благодаря им нейрон имеет сложную форму, перестраивает ее в синаптической части клетки и для внутриклеточного транспорта, в том числе вдоль своей «главной магистрали» — аксона. Нейродегенерации часто сопровождаются образованием агрегатов именно из белков-компонентов клеточного скелета, а нарушение аксонального транспорта — их важная примета. Для ряда болезней характерно разрушение аксонов как таковых или сбои в их работе. Причины не вполне ясны — но ясна роль гиперфосфорилирования промежуточных филаментов в нейронах (нейрофиламентов). Та же модификация стабилизирующего микротрубочки нейронов тау-белка имеет важное значение при болезни Альцгеймера.
Изменения в цитоскелете нейронов нарушают работу синапса (имеющего сложную структуру), энергетический метаболизм и транспорт РНК, а также подстегивают агрегацию белков и гибель нервных клеток.
Патология энергетического метаболизма
Мозг — чрезвычайно «прожорливый» орган в том смысле, что он потребляет очень много энергии. То же можно сказать о нейронах: по уровню потребления АТФ они превосходят другие клетки. Поэтому неудивительно, что нарушения энергетического метаболизма играют важную роль в развитии многих нейродегенеративных заболеваний. Для снабжения нейронов АТФ необходимы молекулы-субстраты — глюкоза или лактат, которые попадают в нервные клетки напрямую из кровотока или транзитом через астроциты.
Сбои в работе митохондрий связаны с недостатком АТФ и нарушением энергозатратных процессов в синапсах и в нейронах в целом. В результате страдают регуляция ионного баланса за счет активного мембранного транспорта, кальциевый гомеостаз, состояние цитоскелета и протеома. Более того, нарушение работы митохондрий чревато окислительным стрессом: дисбалансом между антиоксидантными системами (АОС) и активными формами кислорода (АФК). Те повреждают компоненты клетки: белки, липиды и нуклеиновые кислоты, тем самым запуская нарушения работы нейронов и клеточную смерть.
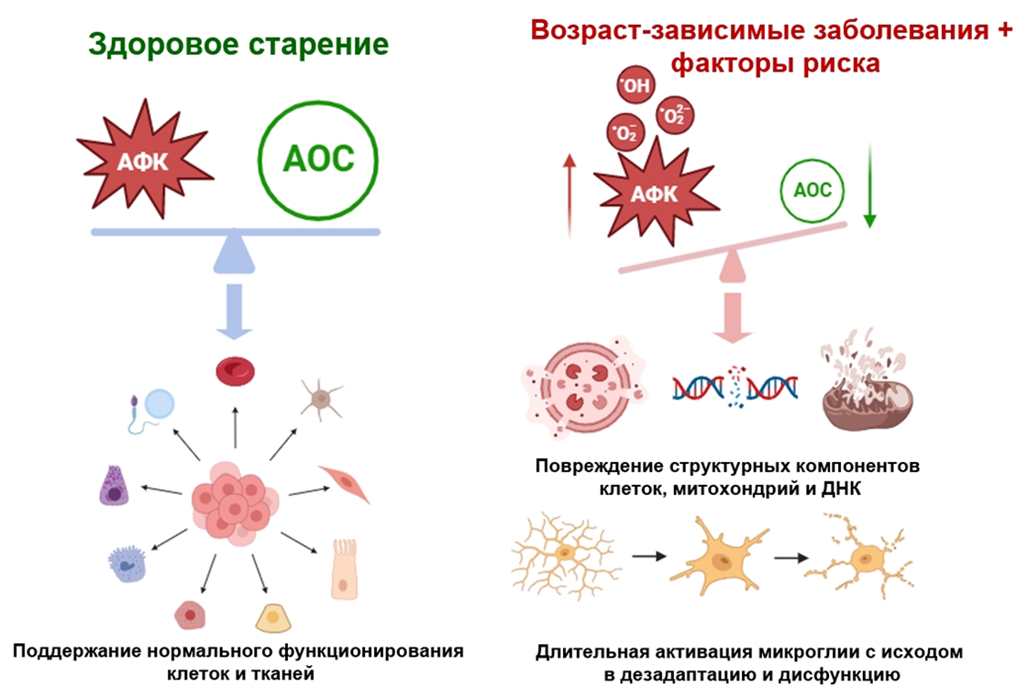
С другой стороны, при повреждении митохондрии — важного депо ионов кальция, — нарушается кальциевый гомеостаз и опосредованная кальцием клеточная сигнализация. Концентрация кальция в клетке растет, что нарушает работу многих ферментов, приводит к высвобождению содержимого лизосом и повреждению белков, ДНК и цитоскелета. Из-за снижения синтеза АТФ становится меньше глутатиона — важного антиоксиданта — и усугубляется окислительный стресс. И вновь мы видим, как многие патологические факторы нейродегенерации связаны и действуют сообща.
Повреждения ДНК и РНК
В ЦНС генотоксичность вызвана прежде всего АФК, которые образуются в митохондриях из-за окислительного фосфорилирования. Кроме того, мутации в генах системы репарации двунитевых разрывов ДНК или системы защиты от репликативного стресса (при атаксии-телеангиэктазии) приводят к атрофии мозговой ткани. Это согласуется с тем, что накопление эндогенных повреждений в ДНК усиливает гибель нервных клеток.
Нарушения метаболизма РНК также участвуют в патогенезе ряда нейродегенераций за счет изменения физиологических процессов, прямой токсичности молекул РНК или стресс-гранул — сгустков из РНК и белка в цитоплазме, — и их участия в агрегации белка.
Отдельный важный механизм, связанный с изменениями ДНК, — это экспансия коротких нуклеотидных повторов, то есть их быстрое разрастание. При этом область повтора не обязательно транслируется с образованием патологического по структуре белка (содержащего полиглутаминовую вставку при одноименных заболеваниях) — их эффект может возникнуть и при попадании в некодирующую или регуляторную область ДНК.
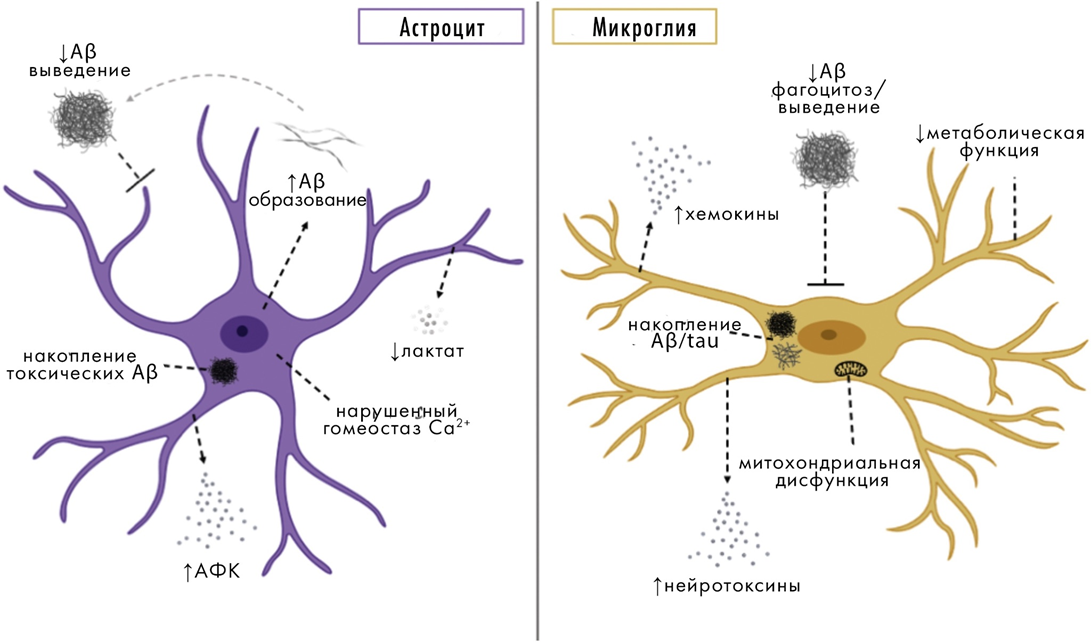
Нейровоспаление
Не застрахован мозг и от воспалительных процессов, которые в этом случае носят специфическое название «нейровоспаление». Оно происходит при участии микроглии и астроцитов. Нейровоспаление очень характерно для нейродегенераций, и его признаки легко различить в мозге при вскрытии умерших пациентов.
Заболеваниям ЦНС, связанным с воспалением, посвящена отдельная статья этого спецпроекта.
Гибель нейронов
Кульминацией совместной разрушительной работы описанных процессов следом за разрушением синапсов и отдельных частей нейронов становится гибель нервных клеток и, как следствие, быстрая атрофия тканей мозга. Перечислим причины, по которым нейроны страдают особенно сильно.
- Нейроны — долгоживущие постмитотические клетки, которые потеряли способность делиться. Их замена возможна только за счет пула стволовых клеток. Как итог, нейроны со временем накапливают повреждения в ДНК, липидах, белках и целых органеллах.
- Повышенная потребность в энергии делает нейроны чувствительными к голоданию — главным образом из-за необходимости поддерживать работу синапсов. Она же означает ускоренную генерацию АФК за счет окислительного фосфорилирования в митохондриях.
- Наличие уязвимых выростов — аксона и дендритов, — для содержания которых необходим клеточный транспорт на значительные расстояния.
- Зависимость от обслуживающих клеток глии, обеспечивающих энергию и защиту.
Такая чувствительность нейронов особенно заметна, когда компенсаторные и защитные механизмы отдельных нейронов и мозга (о которых мы говорили в начале текста) истощаются, и клеткам ничего не остается, кроме как умереть. Сама клеточная гибель может происходить по разным сценариям: лучше прочих описаны механизмы эндогенного и экзогенного апоптоза, а также некроза, хотя клетки могут умирать и за счет других механизмов.