МЕХАНИЗМ ЕСТЬ? А ЕСЛИ НАЙДУ
как они растут /Ещё 5 марта FDA одобрило к применению спрей Spravato с s-кетамином как средство для комбинированной терапии резистентных депрессий. Все, конечно, видели потом эти пресс-релизы про «новый антидепрессант с принципиально новым механизмом действия за последние 30 лет» и что решение FDA чуть ли не первая крупная инновация в современной медицинской практике. Это всё замечательно, знал бы только ещё кто-нибудь этот принципиально новый механизм (равно как, чтобы кто-то определился уже, что считать резистентными и рефрактерными депрессиями, потому что в DSM критериев до сих пор нет, если что).
В целом, вся эта история с «механизмом» получается больше концептуальной штукой (ещё более концептуальной, чем в случае с моноаминами) и упирается в глутаматную гипотезу депрессии. Суть в том, что вот СИОЗ далеко не всегда работают, поэтому в развитии депрессии рассматривают разные другие причины: нарушения циркадных ритмов, дисбаланс моноамины/ацетилхолин, изменения в функциях тироксина и тп. И вот глутаматная гипотеза одно из таких полей для размышлений. У неё любопытная история. Там сначала, в 1990-ом, Trullas and Skolnick на животных моделях показали, что использование конкурентных и неконкурентных антагонистов давало антидепрессант-подобный эффект. И параллельно с этим, тоже в начале 90-ых, в ходе нескольких патологоанатомических исследований было обнаружено, что у самоубийц значительно снижена экспрессия NMDA-рецепторов в нейронах гиппокампа, что говорило о вероятной альтерации глутаматергической системы при жизни.
Это то, с чего глутаматная гипотеза начиналась и на основании чего в 2000 году антидепрессивный эффект кетамина посмотрели в перекрёстном исследовании Вerman at al. на девяти пациентах. Кетамин вводили внутривенно в течение 40 минут в дозировке 0,5мг/кг, аналогично тому, как это делалось в исследовании Krystal (1994, Krystal et al.), использовавшим эту схему в изучении воздействия кетамина на когнитивные и поведенческие функции. И было показано что основной антидепрессивный эффект реализуется не в процессе введения препарата, а начинает определяться приблизительно через 240 минут, в то время как диссоциативное действие заканчивается в течение двух часов после. Антидепрессивный эффект продолжал возрастать в течение нескольких дней после однократного введения, что также давало повод говорить о нём не как как о проявлении возможного интоксикационного синдрома. С этого всё начиналось и на похожих предположениях всё до сих пор и основывается. То есть вот есть кетамин и он работает потому что БЛА-БЛА-БЛА, а не потому что от него штырит (но это не точно).
Кетамин — неконкурентный антагонист NMDA-рецепторов прямого действия. С середины 1960-ых используется в анестезиологической практике, где механизм его действия предельно понятно и подробно расписан (не вдаваясь в подробности, просто напомню, что суть диссоциативной анестезии — угнетение одних участков головного мозга, возбуждение других, появление анальгезирующего эффекта при неполном угнетении сознания и сохранении спонтанного дыхания).
Что касается антидепрессивного действия — всё оооочень размыто. Известно, что субанестетические дозы кетамина ведут к развитию так называемой временной гипофункции NMDA-рецепторов (NRHypo), состоянию, обусловленному частичным блокированием NMDA-рецепторов ГАМКергических нейронов. Ингибирование последних ведёт к так называемому «торможению торможения» (disinhibition). Если грубо, но по делу, ГАМКергические нейроны регулируют (тормозят) два ключевых пути, вовлечённых в процесс: холинергические нейроны первого находятся в базальных отделах переднего мозга, глутаматэргические нейроны второго — в передних ядрах таламуса. И те, и другие оказывают стимулирующее воздействие на пирамидальные нейроны коры. Однако подобный механизм соответствует диссоциативному действию кетамина (если кто-то сможет вдруг мне объяснить чем psychotomimetic отличается от dissociative в контексте употребления кетамина (а не просто клиники), будет вообще классно, пока я нашёл только такое ). Но у нас же концепция, что помогает не потому что «штырит». Здесь начинаются бла-бла-бла.
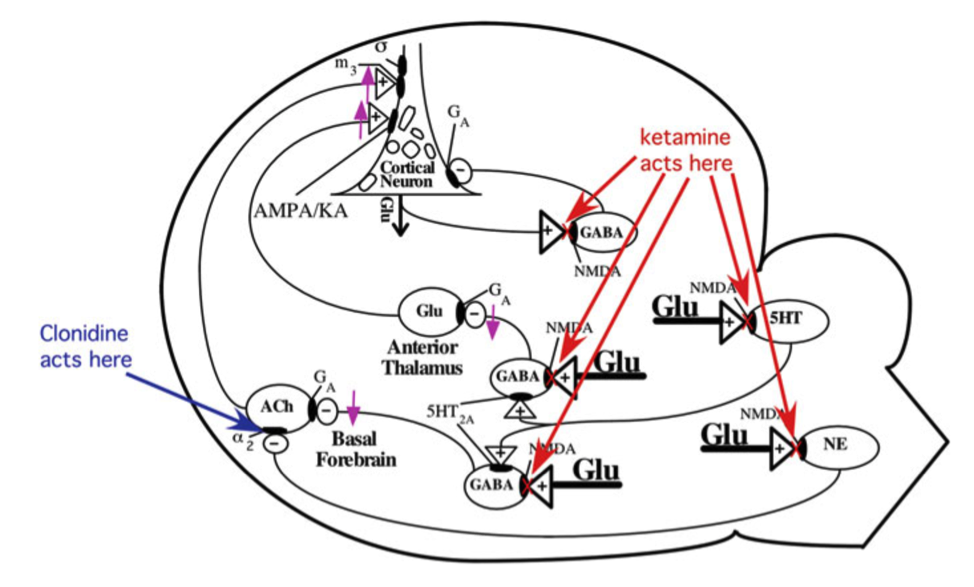
Чтобы объяснить антидепрессивное (отсроченное) действие пытаются рассматривать активацию AMPA-рецепторов, метаболит 2R,6R-гидроксиноркетамин, и активацию мю опиоидных рецепторов. В любом случае всё в идеале стараются привести к синтезу нейротрофического фактора роста (он же BDNF), то есть вписать в контекст неопластической теории, которая сегодня считается наиболее универсальной и давно потеснила моноаминовую «парадигму». Это, к слову, далеко не всегда получается.
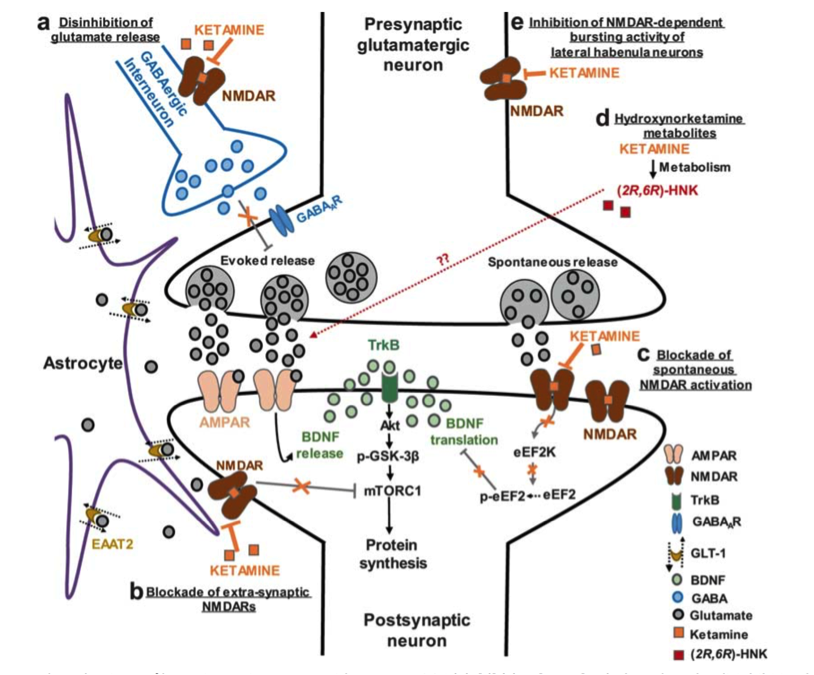
Начинать попытки как-то всё это систематизировать — немношк как лезть в ящик Пандоры, потому что очень-очень много мимолётных, противоречивых данных, что-то вроде: вот мы знаем что S-кетамин in vitro показывает большую аффинность к NMDA рецепторам, чем R-форма и это может обуславливать его более выраженный анальгетический и анестетический (и скорее всего, антидепрессивный) эффекты, и значительно меньше психомиметические свойства. Но вот на животных моделях R-кетамин более эффективен, и вообще, знаете, когда мы говорим что суть антидепрессивного действия сводится к AMPA-рецепторам, мы имеем в виду 2R,6R-гидроксиноркетамин, как метаболит R-кетамина, а как вот это всё привязать к S-кетамину, мы ещё не придумали… (про R и S-формы веществ и препаратов нужен отдельный пост, чуть позже об этом)