Kontrollexperiment Phase 3 - Strukturelle Analyse von Sequenzdaten & genetische Untersuchungen bestätigen: Es gibt keine krankmachenden Viren - TEIL 2 von 2
Corona_Fakten auf Telegram
Im Ersten Teil ging es um die unter der Leitung von Dr. Stefan Lanka durchgeführten Kontrollexperimente, welche bereits im Masernvirusprozess des Jahres 2016 von ihm in Auftrag gegeben worden waren und bei Einhaltung der vorgegebenen wissenschaftlichen Regeln
- A.) Jede wissenschaftliche Behauptung muss überprüfbar, nachvollziehbar und widerlegbar sein.
- B.) Nur, wenn die Widerlegung einer wissenschaftlichen Aussage durch Denkgesetze, Logik und, wenn anwendbar, durch Kontrollexperimente nicht gelungen ist, darf eine Aussage als wissenschaftlich bezeichnet werden.
- C.) Jeder Wissenschaftler ist verpflichtet, seine Aussagen selbst zu überprüfen und zu hinterfragen.
aufzeigten, Dass das eindeutige Ergebnis aus zwei Laboren, die untereinander keinen Kontakt hatten, beweist, dass statt eines Virus typische „Gen-Sequenzen“ aus ganz normalen, gesunden Zellen als zentraler Bestandteil des vermuteten „Masern-Virus“ fehlgedeutet wurden. Damit sind alle Existenzbehauptungen des Masern-Virus widerlegt.
Die Abläufe der Virologie zum Nachweis behaupteter krankmachender Viren ist immer identisch und hat sich im Grunde bis heute nicht verändert.
Die Vorhersage und Schlussfolgerung, dass auch die Kontrollexperimente bei SARS-CoV-2 zu den gleichen Ergebnissen führen werden, ist eine logische Konsequenz.
Wir haben eine Strukturelle Analyse von Sequenzdaten in der Virologie durchgeführt.
Wir untersuchten mit einem einfachen bioinformatischen Protokoll die veröffentlichten Sequenzdaten (BioProject-Zugangsnummer PRJNA603194 in der Datenbank NCBI Sequence Read Archive (SRA)) zur Genomsequenz für SARS-CoV-2 (GenBank: MN908947.3). Die von uns verwendeten Methoden sind nicht spezifisch für SARS-CoV-2 und können ohne besondere Modifikationen auch auf andere Sequenzdaten angewendet werden.
Unsere Ergebnisse zeigen vielfache Missstände in der Behauptung viraler Genomsequenzen auf.
Im Hinblick auf unsere Ergebnisse empfehlen wir neben der Veröffentlichung der endgültigen verwendeten Sequenzdaten stets auch solche, die nur durch die Amplifikation mit zufälligen Hexameren und moderaten Zyklenzahlen entstanden sind, um möglichst unverzerrte Daten zur Strukturanalyse bereitzustellen.
Zusammenfassung
Die de novo meta-transkriptomische Sequenzierung oder vollständige Genomsequenzierung sind in der Virologie anerkannte Methoden zum Nachweis behaupteter pathogener Viren. Dabei werden keine Viruspartikel (Virionen) detektiert und im Sinne des Wortes Isolation, isoliert und biochemisch charakterisiert. Im Falle von SARS-CoV-2 wird oft die gesamte RNA aus Patientenproben (z.B.: bronchoalveoläre Lavageflüssigkeit (BALF) oder Rachen-Nasen-Abstriche) extrahiert und sequenziert. Insbesondere wird nicht der Nachweis erbracht, dass die zur Berechnung der viralen Genomsequenzen verwendeten RNA-Fragmente viralen Ursprungs sind.
Wir untersuchten daher die Publikation „A new coronavirus associated with human respiratory disease in China“ [1] sowie die zugehörigen publizierten Sequenzdaten mit der Bioprojekt-ID PRJNA603194 vom 27.01.2020 zum ursprünglichen Gensequenzvorschlag für SARS-CoV-2 (GenBank: MN908947.3). Eine Wiederholung der de novo Assemblierung mit Megahit (v.1.2.9) zeigte, dass die veröffentlichten Ergebnisse nicht reproduziert werden konnten. Wir detektierten möglicherweise entgegen den Ausführungen in [1] (ribosomale) Ribonukleinsäuren humanen Ursprungs. Weitergehende Analysen lieferten Hinweise auf eine mögliche unspezifische Amplifikation von Lesungen während der PCR-Bestätigung und Bestimmung der Genomtermini, die nicht mit SARS-CoV-2 (MN908947.3) assoziiert sind.
Abschließend führten wir einige referenzbasierte Assemblierungen mit weiteren Genomsequenzen wie beispielsweise SARS-CoV, Humane Immundefizienz-Virus, Hepatitis delta, Masern, Zika, Ebola oder Marburg durch, um die Strukturähnlichkeit der vorliegenden Sequenzdaten mit den jeweiligen Sequenzen zu studieren. Wir erhielten erste Hinweise, dass sich einige der von uns in der vorliegenden Arbeit betrachteten viralen Genomsequenzen aus der RNA unverdächtiger menschlicher Proben gewinnen lassen.
Schlüsselworte
SARS-CoV-2, COVID-19, Virus, De novo Assembly, Ganzgenomsequenzierung, WGS, Bioinformatik, PCR, SARS-CoV, Bat SARS-CoV, Humanes Immundefizienz-Virus, HIV, Hepatitis delta-Virus, Masernvirus, Zika-Virus, Ebola-Virus, Marburg-Virus.
Einführung
Zur Konstruktion viraler Genomsequenzen werden aus verschiedenen Nukleinsäurequellen wie beispielsweise bronchoalveolärer Lavageflüssigkeit (BALF) [1, 2], Nasen-Rachen-Abstrichen [3, 4, 5, 6, 12, 13], Zellkulturbestandteilen bzw. Zellkulturüberständen [2, 11, 12, 13, 14, 16] sowie aus menschlichen [8, 9, 10, 16] und tierischen Proben [7, 15] Nukleinsäuren (RNA oder DNA) isoliert und per Sequenzierung aufgeschlüsselt. Dabei stammen die gewonnenen Nukleinsäuren nicht ausschließlich von zuvor isolierten, also von allem anderen getrennten (Virus-) Partikeln, sondern oft aus der gesamten Probe. Damit ist die Herkunft der zur Berechnung der Genomsequenzen verwendeten Nukleinsäurefragmente a priori ungeklärt.
Im Falle von Ribonukleinsäuren (RNA) wird diese zunächst mit Hilfe RNA-abhängiger DNA-Polymerase in cDNA umgeschrieben. Die DNA oder cDNA wird dann mit Hilfe von Enzymen fragmentiert und per Polymerase Kettenreaktion (PCR) vermehrt, bevor die eigentliche Sequenzierung, also die Bestimmung der Nukleotidabfolge der kurzen DNA bzw. cDNA-Fragmente, erfolgt. Bei der Amplifikation werden neben zufälligen Primersequenzen (zufällige Hexameren) auch in Abhängigkeit der betrachteten Referenz- oder Zielgenome hochspezifische Primersequenzen verwendet [z.B.: 1, 3, 4, 5, 6, 7, 8, 17, 18]. Die so gewonnenen Sequenzdaten werden schließlich mit bioinformatischen Algorithmen verarbeitet
Zwei verbreitete Methoden zur Ermittlung viraler Genomsequenzen stellen die de novo meta-transkriptomische Assemblierung [1, 12] und die vollständige Genomsequenzierung [3, 4, 5, 6, 17, 18] dar. Während bei der de novo meta-transkriptomischen Assemblierung oft keine bzw. nur nachgelagert Referenzsequenzen Verwendung finden, werden bei der vollständigen Genomsequenzierung eine Vielzahl spezifischer Primersequenzen herangezogen, die bereits teilweise zusammen 4% bis 17% des Zielgenoms abdecken [1, 17]. Zur Amplifikation der cDNA kommen oft 35 bis 45 Zyklen [1, 6, 17] zur Anwendung.
Im Falle von SARS-CoV-2 (GenBank: MN908947.3) [1] wurde der Virusgenomsequenzvorschlag durch de novo meta-transkriptomische Assemblierung der RNA aus der BALF eines Patienten in Wuhan, China berechnet. Zur Assemblierung der Contigs wurden die Assembler Megahit (v.1.1.3) und Trinity (v.2.5.1) verwendet. Megahit generierte insgesamt 384.096 (200 nt – 30.474 nt) und Trinity berechnete 1.329.960 (201 nt – 11.760 nt) Contigs. Bemerkenswert sind die großen Unterschiede beider Assemblierungen. Das längste Contig, zusammengesetzt mit Megahit, zeigte nach [1] eine hohe Überstimmung (89,1%) mit dem Genom bat SL-CoVZC45 (Genbank: MG772933) und wurde zum Design der Primer für die PCR-Bestätigung und zur Bestimmung der Genomtermini verwendet.
Die virale Genomorganisation wurde durch Sequenzausrichtung auf zwei repräsentative Spezies der Gattung Betacoronavirus bestimmt, ein mit Menschen assoziiertes Coronavirus (SARS-CoV Tor 2, GenBank: AY274119) und ein mit Fledermäusen assoziiertes Coronavirus (bat SL-CoVZC45, GenBank: MG772933).
Aus der Patientenprobe wurde dabei kein pathogenes Viruspartikel identifiziert und biochemisch charakterisiert, welches eindeutig mit der Sequenz MN908947.3 assoziiert ist. Vielmehr wurde die gesamte RNA aus der BALF eines Patienten extrahiert und verarbeitet. Es fehlt der Nachweis, dass zur Konstruktion des behaupteten Virusgenoms für SARS-CoV-2 ausschließlich virale Nukleinsäuren verwendet wurden. Weiter wurden in Bezug auf die Konstruktion des behaupteten viralen Erbgutstranges keine Ergebnisse möglicher Kontrollexperimente veröffentlicht [Auch per schriftlicher Anfrage übermittelte Prof. Zhang keine Kontrollversuche]. Dies gilt ebenso für alle weiteren in der vorliegenden Arbeit betrachteten Referenzsequenzen. Im Falle von SARS-CoV-2 bestünde eine naheliegende Kontrolle darin, dass aus unverdächtigen RNA-Quellen menschlichen, oder auch anderen Ursprungs, das behauptete Virusgenom nicht assembliert werden kann.
In der vorliegenden Publikation untersuchten wir die Reproduzierbarkeit von de novo Assemblierungen anhand der veröffentlichten Original-Sequenzdaten zur ursprünglichen Arbeit zum Coronavirus SARS-CoV-2 [1]. Weiter untersuchten wir die Strukturähnlichkeit der vorliegenden Sequenzdaten mit weiteren öffentlich zugänglichen viralen Referenzsequenzen für (Fledermaus-) SARS-CoV [1, 7, 13, 14], Humane Immundefizienz-Virus [8], Hepatitis delta [9], Masern [11, 12], Zika [10], Ebola [15], Marburg [16] (Tabellen und Abbildungen: Tabelle 3). Dazu stellen wir hier ein einfaches bioinformatisches Protokoll vor. Zur Absicherung unserer Ergebnisse betrachteten wir auch zufällig generierte und fiktive Genomsequenzen, um eine reine Zufälligkeit unserer Resultate auszuschließen.
Hauptteil
Erneute de novo Assemblierung der veröffentlichten Sequenzdaten
Zur Wiederholung der de novo Assemblierung haben wir die ursprünglichen Sequenzdaten (SRR10971381) vom 27.01.2020 am 30.11.2021 unter Verwendung der SRA-Tools [19] aus dem Internet heruntergeladen. Zur Vorbereitung der gepaarten Sequenzlesungen für den eigentlichen Assemblierungsschritt mit Megahit (v.1.2.9) [20] verwendeten wir den FASTQ-Präprozessor fastp (v.0.23.1) [21]. Nach der Filterung der paired-end-Lesungen verblieben 26.108.482 von den ursprünglich insgesamt 56.565.928 Lesungen mit einer Länge von etwa 150 bp. Ein großer Teil der Sequenzen, vermutlich mehrheitlich solchen menschlichen Ursprungs wurden von den Autoren mit „N“ für unbekannt überschrieben und daher von fastp herausgefiltert. Dies ist im Sinne der Wissenschaftlichkeit als problematisch anzusehen, da nicht mehr alle Schritte nachvollzogen bzw. reproduziert werden können. Zur aufwendigen Contigerstellung aus den verbliebenen kurzen Sequenzlesungen verwendeten wir Megahit (v.1.2.9) in der Standardeinstellung.
Wir erhielten mit 28.459 (200 nt – 29.802 nt) Contigs deutlich weniger als in [1] beschrieben. Abweichend von den Darstellungen in [1] umfasste das von uns längste zusammengesetzte Contig lediglich 29.802 nt und damit 672 nt weniger als das längste Contig mit 30.474 nt, welches nach [1] nahezu das gesamte virale Genom umfasste. Unser längstes Contig zeigte auf einer Länge von 29.801 nt eine perfekte Übereinstimmung mit der Sequenz MN908947.3 (Tabellen und Abbildungen, Tabellen 1, 2).
Damit konnten wir das für die wissenschaftliche Überprüfung so wichtige längste Contig mit 30.474 nt nicht reproduzieren. Folglich kann es sich bei den veröffentlichten Sequenzdaten nicht um die ursprünglichen zur Assemblierung verwendeten Lesungen handeln.
Nach der Assemblierung der Contigs ermittelten wir durch Mapping der kurzen Sequenzen an die 28.459 ermittelten Contigs mit Bowtie2 (v.2.4.4) [22] die jeweilige Abdeckungsfülle. Anschließend glichen wir die 50 Contigs mit der größten Abdeckungsfülle sowie die 50 längsten Contigs am 05.12.2021 bzw. 20.12.2021 mit der Nukleotiddatenbank (Blastn) ab. Die detaillierten Abfrageergebnisse können den Tabellen und Abbildungen: Tabellen 1, 2 entnommen werden.
Ein Vergleich unserer Ergebnisse (Tabellen und Abbildungen: Tabelle 1) mit denen aus [1, ergänzende Tabelle 1] zeigen bemerkenswerte Unterschiede. Im Folgenden wird an die Contig-Id‘s aus [1] zur besseren Abgrenzung zu unseren Contig-Id’s „1_“ vorangestellt. Ganz allgemein lässt sich feststellen, dass unsere Abfragetreffer bzgl. der Akzessionsnummern nicht exakt mit denen aus [1] übereinstimmen. In Bezug auf die Subjektbeschreibungen beobachteten wird weitestgehend eine gute Übereinstimmung. Weiter zeigte sich, dass unsere Contigs, mit Ausnahme des längsten Contigs (1_k141_275316), eine größere Länge und tendenziell eine größere Fülle der Abdeckung aufweisen. Eindeutig ist der Fall bei Contig 1_k141_179411 im Vergleich zum Contig k141_12253. Das erstgenannte besitzt eine Länge von 2.733 nt, während das zweitgenannte 5.414 nt lang ist. Dies gibt einen ersten möglichen Hinweis darauf, dass es während der PCR-Bestätigung mit den für MN908947.3 aus 1_ k141_275316 konstruierten Primern zu einer unspezifischen Amplifikation von nicht mit SARS-CoV-2 assoziierten Sequenzlesungen kam.
An dieser Stelle soll noch detailliert auf das Contig mit der Identifikation k141_27232 eingegangen werden, mit welchem 1.407.705 Sequenzen assoziiert sind, und damit etwa 5% der verbliebenen 26.108.482 Sequenzen. Der Abgleich mit der Nukleotiddatenbank am 05.12.2021 zeigte eine hohe Übereinstimmung (98,85%) mit „Homo sapiens RNA, 45S pre-ribosomal N4 (RNA45SN4), ribosomal RNA“ (GenBank: NR_146117.1, vom 04.07.2020). Diese Beobachtung steht im Widerspruch zu der Behauptung in [1], dass eine ribosomale RNA-Depletion durchgeführt und humane Sequenzlesungen unter Verwendung des menschlichen Referenzgenoms (human release 32, GRCh38.p13) gefiltert wurden. Bemerkenswert hierbei ist die Tatsache, dass die Sequenz NR_146117.1 erst nach der Veröffentlichung der hier betrachteten Sequenzbibliothek SRR10971381 publiziert wurde.
Diese Beobachtung unterstreicht die Schwierigkeit der a priori Bestimmung des exakten Ursprungs der einzelnen Nukleinsäurefragmente, die zur Konstruktion behaupteter viraler Genomsequenzen Verwendung finden.
Referenzbasierte Sequenzstrukturanalyse
Grundsätzlich haben wir die paired-end-Lesungen (2x151 bp) mit BBMap [23] an die von uns betrachteten Referenzsequenzen (Tabellen und Abbildungen: Tabelle 3) unter Verwendung relativ unspezifischer Einstellungen gemappt. Anschließend variierten wir die Mindestlänge (M1) und die Mindest(nukleotid)identität (M2) mit reformat.sh um entsprechende Teilmengen der zuvor gemappten Sequenzen mit entsprechender Güte zu erhalten. Die Erhöhung von Mindestlänge M1 oder Mindestnukleotididentität M2 erhöht dabei die Signifikanz des jeweiligen Mappings. Anschließend bildeten wir mit den jeweiligen Teilmengen von gewählter Güte Konsensussequenzen bzgl. der jeweils betrachteten Referenz. Dabei setzten wir alle Basen mit einer Qualität geringer als 20 auf „N“ (unbekannt). Eine Qualität von 20 bedeutet dabei eine Fehlerrate von 1% je Nukleotid, was im Rahmen unserer Analysen als ausreichend angesehen werden kann. Schließlich erfolgte die Beurteilung der Übereinstimmung von Referenz- und Konsensussequenzen mit BWA [24], Samtools [25] und Tablet [26]. Das geordnete Paar (M1; M2) = (37; 0,6) wurde gerade so gewählt, dass sich für die Referenz LC312715.1 Fehlerraten F1 bzw. F2 von kleiner 10% ergaben. Die Ergebnisse aller durchgeführten Berechnungen sind in Tabellen und Abbildungen: Tabelle 4 dargestellt.
Die Berechnungen zeigen die höchste Signifikanz für die Wahl des geordneten Paares (37; 0,6), was an den jeweils höchsten Fehlerraten zu erkennen ist. Eine vergleichbare Signifikanz liefern die geordneten Paare (47; 0,50) und (25; 0,62). Während die mit Coronaviren assoziierten Genomsequenzen in etwa Fehlerraten von über 10% für alle betrachteten geordneten Paare (M1; M2) zeigen, liegen die Fehlerraten der beiden Sequenzen LC312715.1 (HIV) und NC_001653.2 (Hepatitis delta) unter 10% und nehmen bei den geordneten Paaren (32; 0,60) und (30; 0,60) weiter ab. Die Sequenz MG772933_short besteht im Wesentlichen aus dem Teil, welcher nicht mit den SARS-CoV-2 assoziierten Lesungen abdeckbar ist (siehe Tabellen und Abbildungen: Abbildung 3). Auch hier konnte keine Verbesserung durch Reduktion der Werte für M1 und M2 erreicht werden. Die Fehlerraten für die Sequenzen NC_039345.1 (Ebola), NC_024781.1 (Marburg), AF266291.1 und KJ410048.1 (Masern) liegen deutlich über denen von LC312715.1 und NC_001653.2.
Während die Nukleinsäuresequenzen zur Errechnung der erstgenannten Genome in Vero Zellen propagiert wurden, entstammen die für LC312715.1 und NC_001653.2 verwendeten Nukleinsäuresequenzen direkt aus Proben menschlichen Ursprungs (Tabellen und Abbildungen: Tabelle 3).
Es stellt sich daher die Frage, ob dieses Resultat auf Strukturunterschiede der jeweiligen Nukleinsäurequellen oder auf die jeweils verwendeten Sequenzierungsprotokolle zurückzuführen ist. So könnten möglicherweise die zur Umwandlung der RNA in cDNA verwendete reverse Transkriptase oder die zur Amplifikation herangezogenen Primersequenzen sowie die Amplifikationszyklen zu Unterschieden der gewonnenen Sequenzbibliotheken führen.
Die höchsten Fehlerraten F1 und F2 zeigen die zufällig generierten fiktiven Genomsequenzen rnd_uniform, rnd_wuhan, rnd_wh_mk_1 und rnd_ wh_mk_2, sodass die hier gefundenen Ergebnisse nicht rein zufällig sind.
Grafische Analyse der Abdeckungsverteilungen und Leselängen
Nachdem wir die Möglichkeit zur Bildung von Konsensussequenzen mit hoher Güte bzgl. einiger Referenzsequenzen beobachtet haben, analysierten wir die Abdeckungsverteilung der assoziierten kurzen Sequenzlesungen (Tabellen und Abbildungen: Abbildungen 1-22) sowie die Verteilung der Leselängen (Tabellen und Abbildungen: Abbildungen 23-25). Dazu haben wir zuvor die kurzen Sequenzlesungen mit BBMap an die jeweiligen Referenzsequenzen gemappt ((M1; M2) = (37; 0,60)). Neben den kurzen Sequenzen, haben wir ebenfalls die 26 Primerpaare [1, Supplementary Table 8. PCR primers used in this study.] zur Gesamtgenomsequenzierung von SARS-CoV-2 (GenBank: MN908947.3) an die betrachteten Referenzgenome gemappt.
Die anschließende Analyse erfolgte via Tablet und dem Tabellenkalkulationsprogramm Excel. Zunächst betrachten wir die zufällig generierte Referenz rnd_uniform
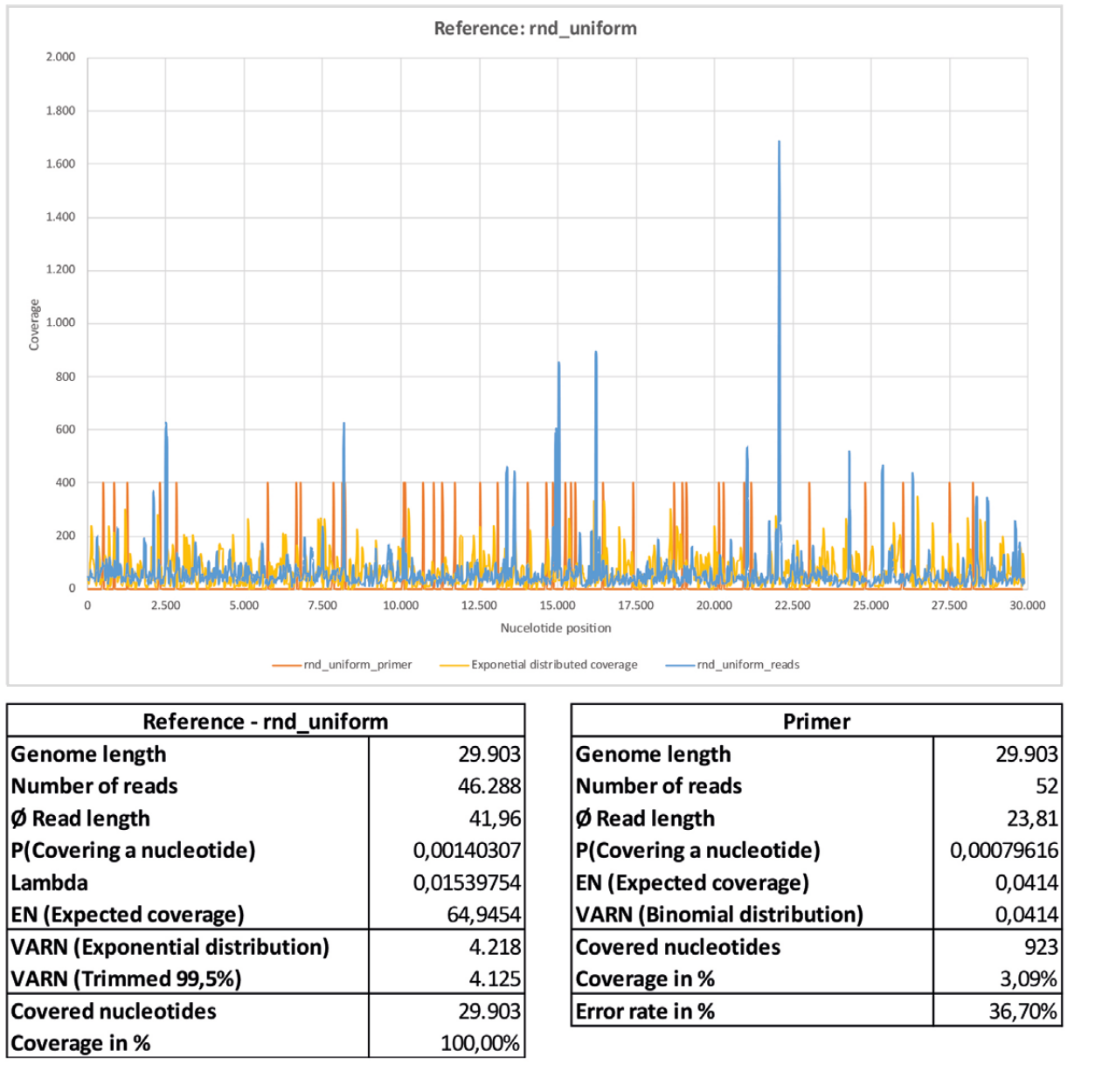
Abbildung 13: Referenz rnd_uniform.
a) rnd_uniform_reads gemappt mit BBMap (M1; M2) = (37; 0,60).
b) rnd_uniform_primer gemappt mit BBMap.
c) Exponential distributed coverage wurde durch stoch astische Simulation mit Hilfe der Inversionsmethode erzeugt.
d) Die 26 Primerpaare ([1, Supplementary Table 8. PCR primers used in this study.]) verteilen sich ungleichmäßig über das gesamte Referenzgenom hinweg. Die Primerpositionen korrelieren nur schwach mit Bereichen hoher Nukleotidabdeckung, wobei diese jeweils nur wenige Nukleotide umfassen.
e) Die Verteilung von rnd_uniform_reads erscheint weitestgehend zufällig. Die Varianz der betrachteten Exponentialverteilung stimmt gut mit der getrimmten empirischen Varianz überein.
Die Abdeckung (rnd_uniform_reads) schwankt zufällig und relativ homogen über alle Nukleotidpositionen hinweg. Die Struktur ist vergleichbar mit der zufällig generierten Abdeckung (Exponential distributed coverage), wobei die Varianz etwas geringer erscheint. An wenigen vereinzelten Nukleotidpositionen zeigt die Abdeckung im Vergleich zum Durchschnitt hohe Abdeckungen, die sich aber jeweils nur über wenige zusammenhängende Nukleotidbereiche erstrecken. Ein Zusammenhang mit den Primerpositionen ist nur schwach ausgeprägt zu erkennen. Die rein zufällig erscheinende Abdeckung mit den kurzen Sequenzlesungen korreliert mit einer nicht durchgängig mappbaren Konsensussequenz und hoher Fehlerrate F1 von 38,60%. Die zufällige (innere) Nukleotidstruktur der stochastisch simulierten Referenzsequenz „rnd_uniform“ ist in den hier untersuchten Sequenzdaten somit eher nicht enthalten.
Im Kontrast dazu betrachten wird nun das Referenzgenom für SARS-CoV-2 (GenBank: MN908947.3)
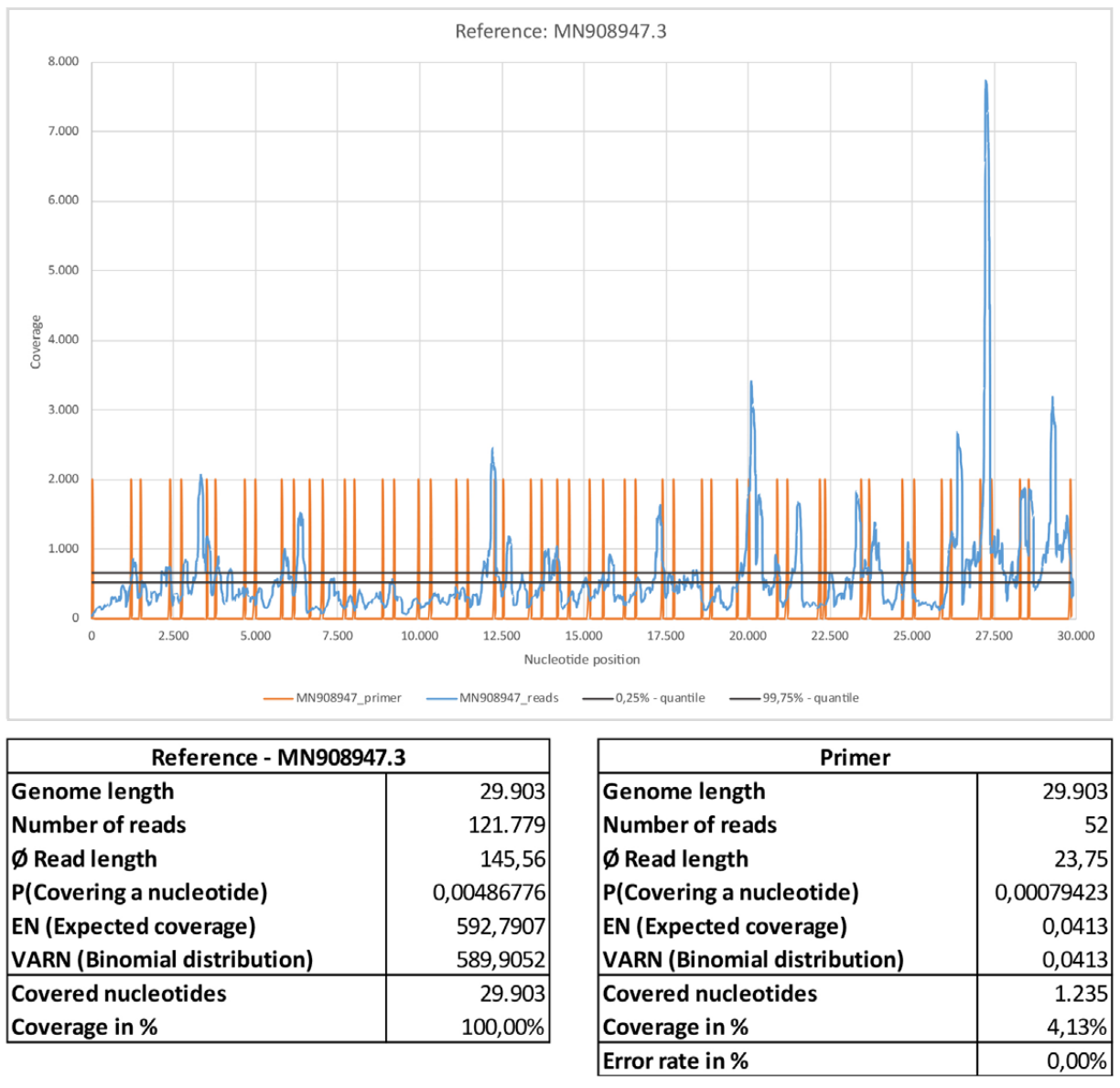
Abbildung 1: Referenz MN908947.3.
a) MN908947.3_reads gemappt mit Bowtie2 unter Verwendung der Standardeinstellungen.
b) MN908947.3_primer gemappt mit BBMap.
c) Quantile wurden ermittelt aus EN und VARN unter der Verteilungshypothese einer Binomialverteilung.
d) Die 26 Primerpaare ([1], Supplementary Table 8. PCR primers used in this study.) liegen gleichmäßig verteilt über das gesamte Referenzgenom hinweg. Die Primerpositionen korrelieren mit Bereichen hoher Nukleotidabdeckung
Im Unterschied zu Abbildung 13 zeigt die Abdeckungsverteilung eher ein Wellenmuster mit regelmäßigen deutlich erhöhten Nukleotidabdeckungen. Die 26 Primerpaare verteilen sich gleichmäßig über alle Nukleotidpositionen der Referenzsequenz. In der Nähe von Nukleotidpositionen mit einer im Vergleich zum Durchschnitt hohen Nukleotidabdeckung befinden sich oft auch die Primerpositionen. Dies zeigt, dass nicht alle Teile des Referenzgenoms gleichmäßig amplifiziert wurden. Unter der Annahme, dass alle 29.903 Nukleotidpositionen bei SARS-CoV-2 assoziierten Lesungen mit gleicher Wahrscheinlichkeit vorkommen, müsste die Abdeckung für jede Nukleotidposition mit 99,5% Wahrscheinlichkeit zwischen den beiden Linien liegen. Dies ist bei etwa 90% der Nukleotidpositionen nicht der Fall. A priori würde man erwarten, dass, wenn hinreichend Virus-RNA in der Probe vorliegt und hinreichend viele Sequenzstückchen gelesen werden, eine homogene Abdeckung der Nukleotide innerhalb des Virusgenoms erreicht würde.
Die nachfolgende Grafik ermöglicht das Studium der Verteilungen der Leselängen der soeben betrachteten Referenzen (rnd_uniform und MN908947.3)
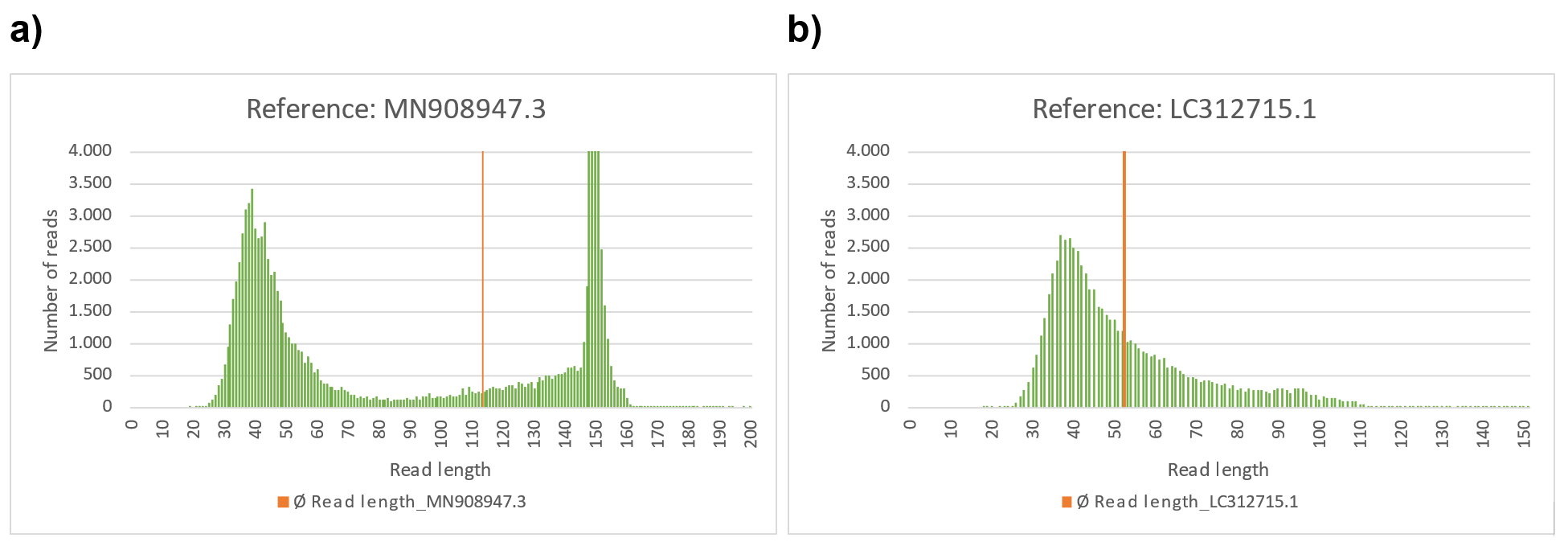
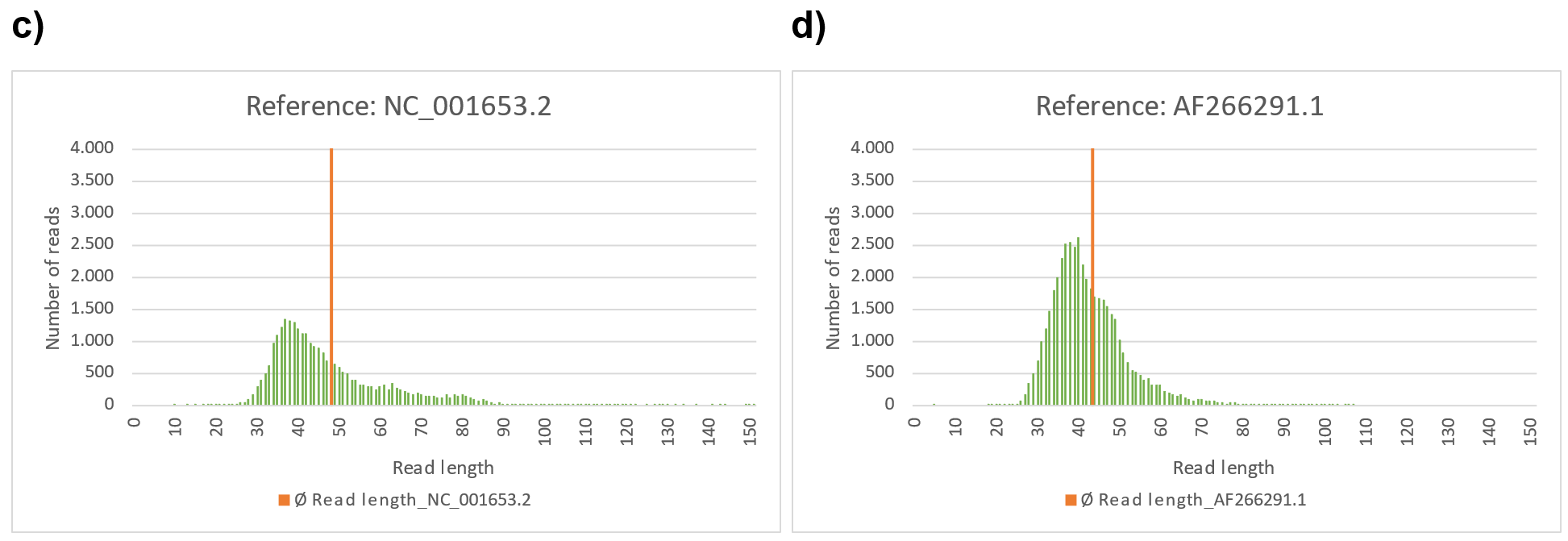
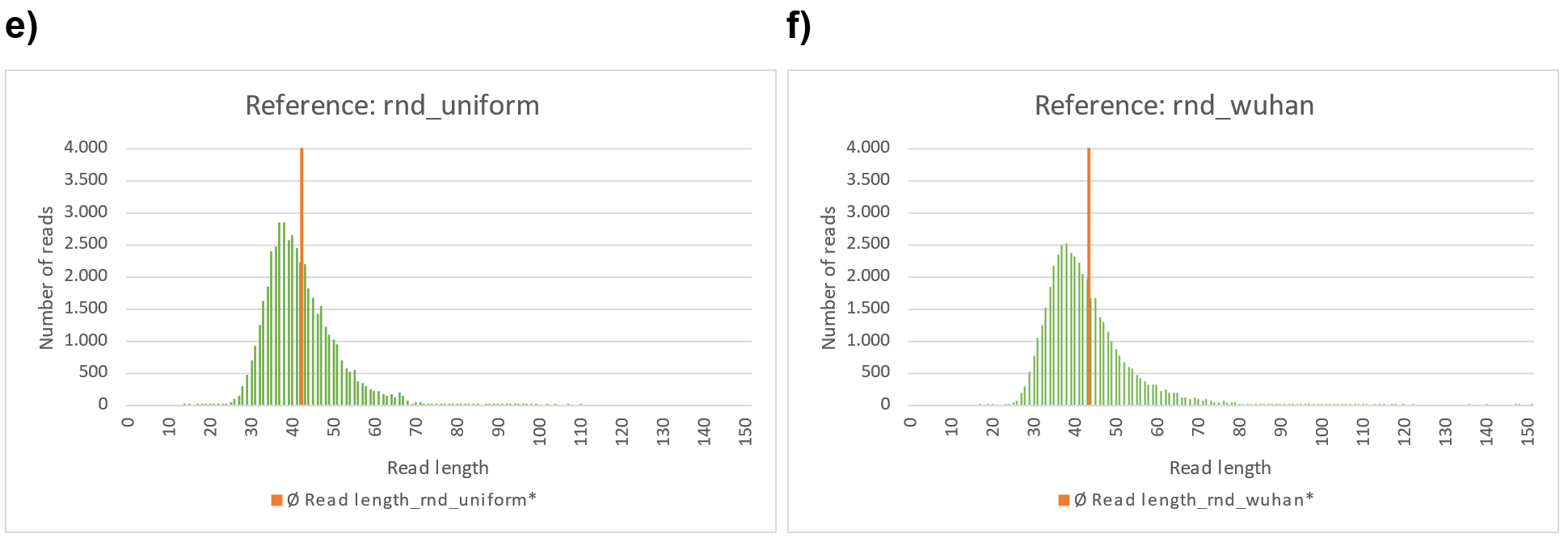
Abbildung 23: a)-f) Mapping mit BBMap (M1; M2) = (37; 0,60). Analyse in Excel.
Abbildung 23e) zeigt die Verteilung der Leselängen im Falle der Referenz „rnd_uniform“. Die durchschnittliche Leselänge liegt bei 41,96 nt nur leicht rechts neben dem Maximum der Verteilung. Im Vergleich dazu zeigt die Verteilung für die Referenz MN908947.3, Abbildung 23a) einen markanten zu Abbildung 23e) ähnlichen (zufälligen) Bereich und einen ausgeprägten Bereich mit Lesungen von etwa 150 nt Länge. Die durchschnittliche Leselänge liegt bei über 110 nt. Alle Referenzsequenzen mit einer vergleichbaren und daher eher zufälligen Verteilung der Leselängen wie bei der stochastisch simulierten Referenz „rnd_uniform“ (Tabellen und Abbildungen: Abbildung 23d), f); Abbildung 24d), e), f); Abbildung 25a)-c)) zeigen auch hohe Fehlerraten F1 und F2 (Tabellen und Abbildungen: Tabelle 4)
Diese Feststellung wird durch die nachfolgende Analyse unterstrichen. Um die innere Struktur der veröffentlichten ca. 56 Mio. Sequenzen besser zu verstehen, haben wir für die Sequenz MN908947.3 bei der Teilmengenbildung im Anschluss an das Mapping mit BBMap neben M1 und M2 die zusätzliche Bedingung maxlength=100 berücksichtigt.

Abbildung 2: Referenz MN908947.3.
a) MN908947.3_reads gemappt mit Bowtie2 unter Verwendung der Standardeinstellungen.
b) MN908947.3_short_reads gemappt mit BBMap (M1; M2) = (37 (max. 100); 0,60).
c) Exponential distributed coverage wurde durch stochastische Simulation mit Hilfe der Inversionsmethode erzeugt. Die Abdeckungsverteilung MN908947.3_short_reads zeigt ein eher zufälliges Muster, weist aber eine höhere Varianz (nach Kappung der 0,5% höchsten Nukleotidabdeckungen. Die ist im Wesentlichen auf die wenigen Ausschläger Abdeckungsverteilung zu erklären.
Durch Ausschluss aller mappbaren Sequenzen mit einer Länge oberhalb von 100 Nukleotiden, wurden im Wesentlichen die etwa 120.000 mit SARS-CoV-2 assoziierten Lesungen entfernt. Die Abdeckungsverteilung der verbleibenden kurzen Sequenzen erscheint analog zu Abbildung 13 nunmehr zufällig. Erneut korreliert dies mit hohen Fehlerraten R1 (29,90%) und R2 (29,96%). Dies zeigt, dass in den veröffentlichten Sequenzen, mit Ausnahme der etwa 120.000 (Tabellen und Abbildungen. Tabelle 1) assoziierten kurzen Lesungen keine nennenswerte Struktur der Referenz MN908947.3 enthalten ist.
Bevor wir auf einige von uns untersuchte Referenzgenome detailliert eingehen, möchten wir zunächst die Abdeckung zweier weiterer Contigs betrachten (k141_12253 und k141_20796). Während sich das Contig mit der Identifikation k141_12253 durch eine relativ hohe Abdeckung auszeichnet, gehört k141_20796 zu den drei längsten errechneten Contigs.
Abbildung 18: Referenz k141_12253.
a) k141_12253_reads gemappt mit Bowtie2 unter Verwendung der Standardeinstellungen.
b) k141_12253_primer gemappt mit BBMap.
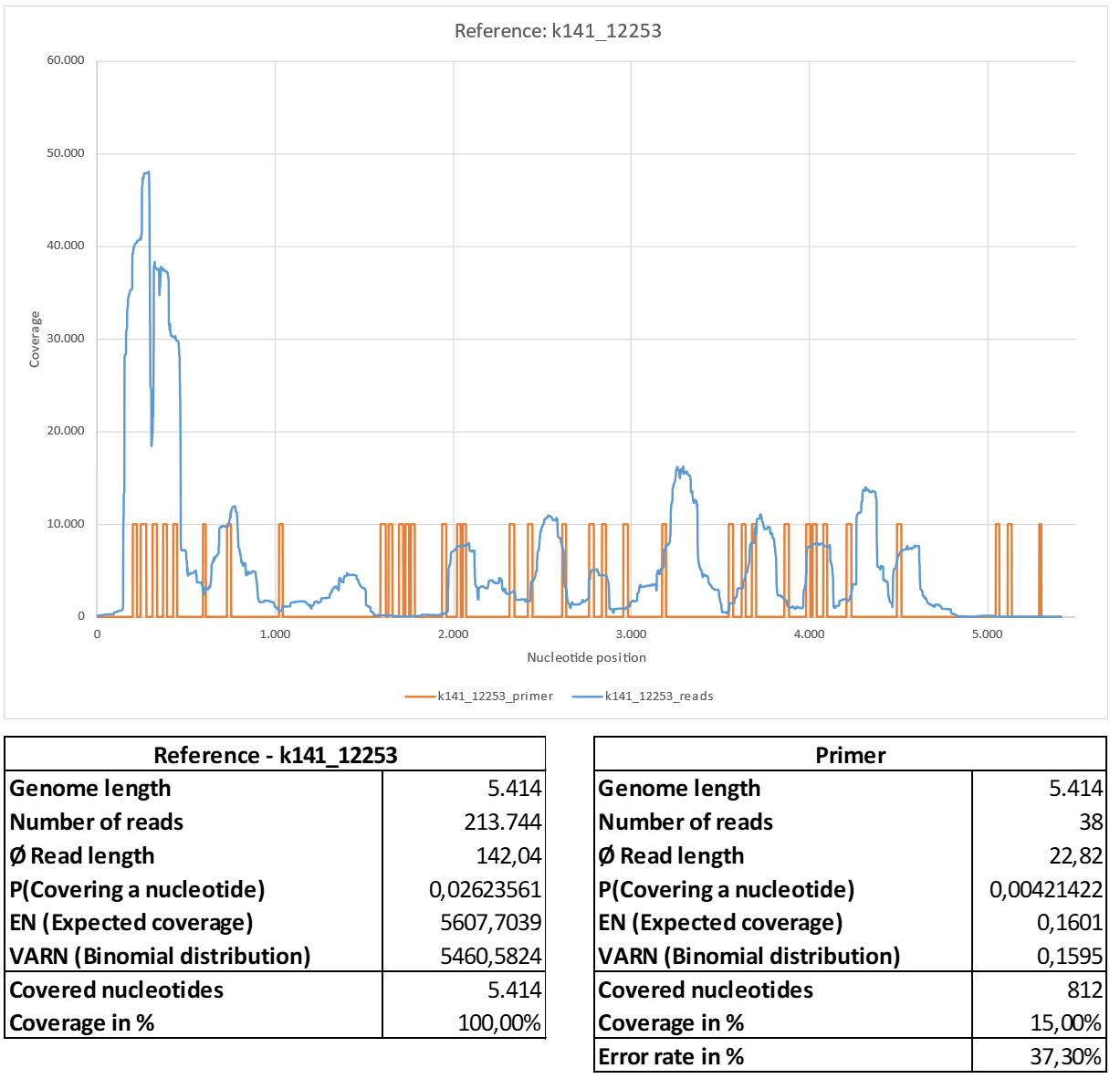
Das Contig k141_12253 zeigt eine hohe Ähnlichkeit mit dem Bakterium Leptotrichia (GenBank: CP012410.1). Von den 52 veröffentlichen Primersequenzen konnten 38 mit einer relativ hohen Fehlerrate von 37,30% an die Referenz k141_12253 gemappt werden. Die Abdeckungsverteilung stellt sich äußerst inhomogen dar und zeigt insbesondere innerhalb der ersten 500 Nukleotide eine im Vergleich zum Durchschnitt extrem hohe Nukleotidabdeckung.
Die Bereiche mit einer hohen Abdeckung korrelieren dabei mit den ermittelten Primerpositionen. Dies könnte darauf hindeuten, dass nicht ausschließlich SARS-CoV-2 assoziierte Lesungen in großer Menge amplifiziert wurden. Unter Berücksichtigung der relativ hohen Fehlerrate von 37,30% würde dies eine relativ unspezifische Amplifikation bedeuten. So stellt sich die Frage, ob durch die Amplifikation der cDNA mit den spezifischen Primersequenzen gewonnenen Lesungen bereits in der Ausgangsprobe enthalten waren, oder durch das Verfahren selbst entstanden sind.
Abbildung 21: Referenz k141_20796.
a) k141_20796_reads gemappt mit Bowtie2 unter Verwendung der Standardeinstellungen.
b) k141_20796_primer gemappt mit BBMap.
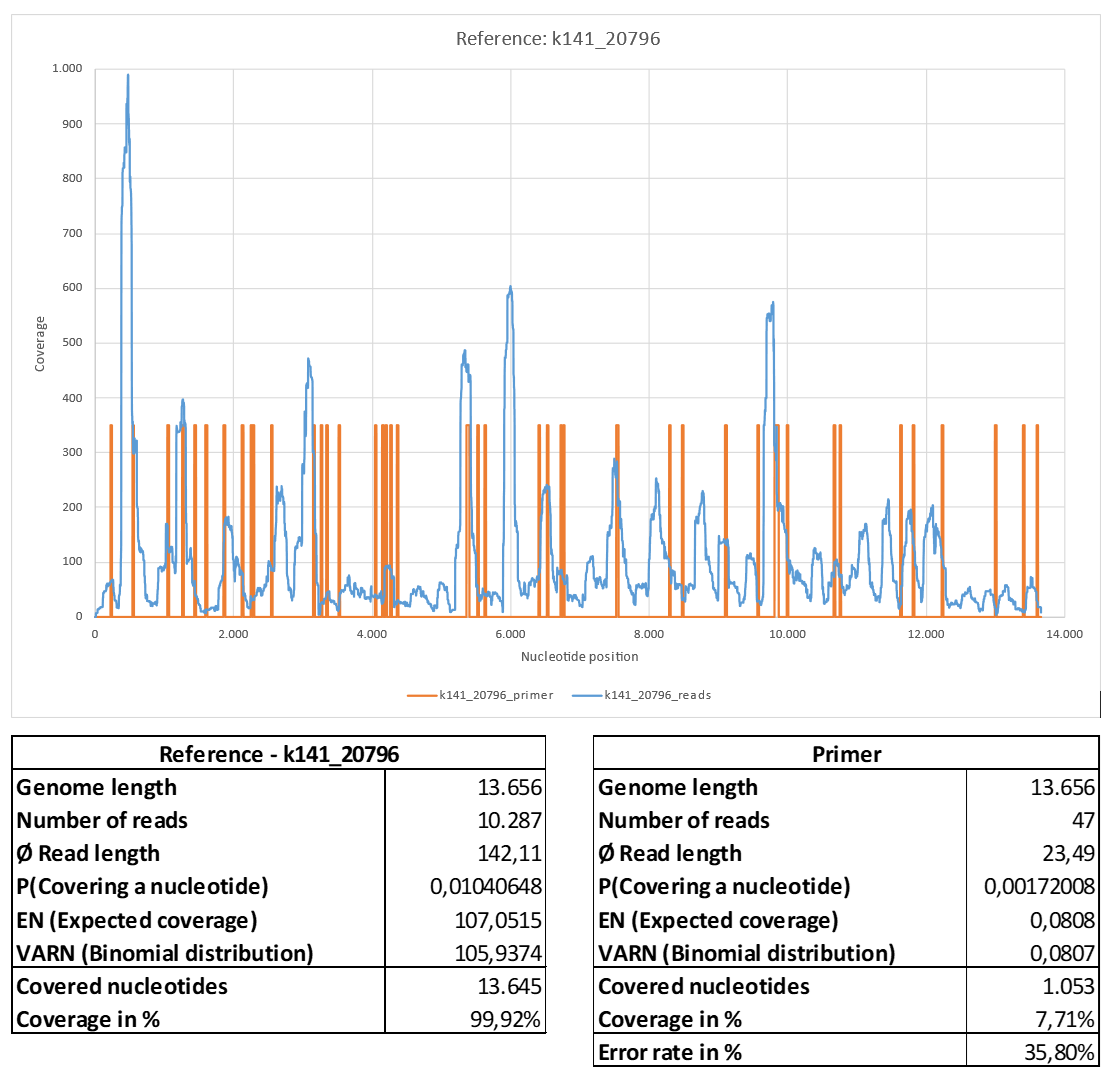
Das Contig k141_20796, welches eine hohe Übereinstimmung mit dem Bakterium Veillonella parvula (GenBank: LR778174.1) aufweist, zeigt eine geringere Abdeckung mit assoziierten Lesungen im Vergleich zum Contig mit der Identifikation k141_12253. Die Struktur der Nukleotidabdeckung ähnelt der von SARS-CoV-2 (GenBank: MN908947.3). Insbesondere ist die Abdeckung erneut inhomogen, was auf eine ungleichmäßige Amplifikation hinweist.
Aufgrund der höheren Nukleotidlänge, konnten nun 47 der 52 veröffentlichten Primersequenzen mit einer mittleren Fehlerrate von 35,8% an das Referenzcontig gemappt werden. Erneut korrelieren die Primerpositionen gut mit den Bereichen einer hohen Nukleotidabdeckung. Dies könnte auch hier auf eine unspezifische Amplifikation von Sequenzen, welche nicht mit SARS-CoV-2 (GenBank: MN908947.3) assoziiert sind, hindeuten.
In dem vorliegenden Abschnitt wollen wir noch detaillierter auf die Referenzsequenzen „Human immunodeficiency virus 1“ (GenBank: LC312715.1) und „Measles virus genotype D8 strain MVi/Mu enchen“ (GenBank: KJ410048.1) eingehen. Alle übrigen Abbildungen können den ergänzenden Materialien (Tabellen und Abbildungen: Abbildungen 1-22 und Abbildungen 23-25) entnommen werden.
Abbildung 6: Referenz LC312715.1.
a) LC312715.1_short_reads gemappt BBMap (M1; M2) = (37; 0,60).
b) LC312715.1_primer gemappt mit BBMap
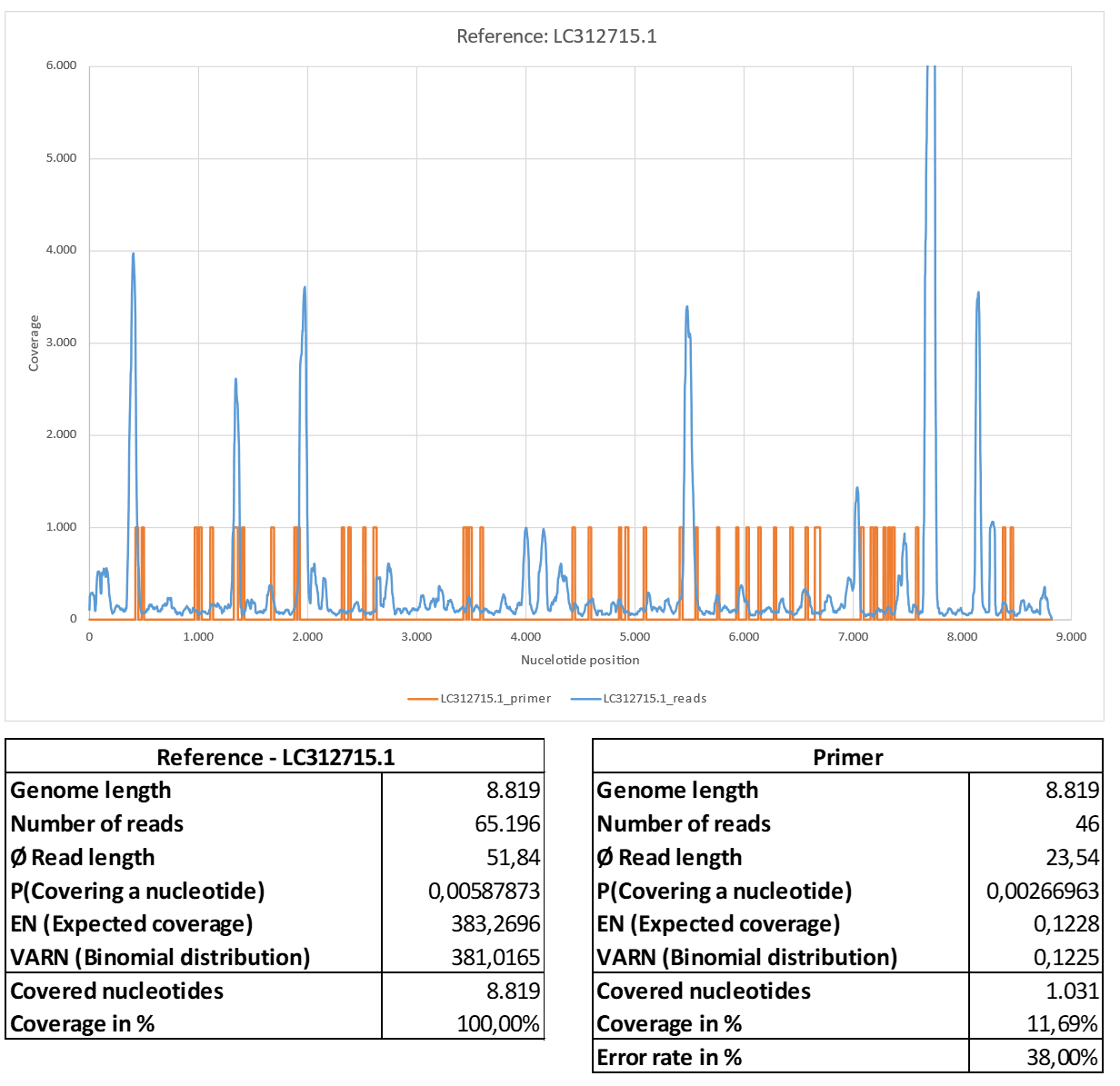
Bereits im vorherigen Abschnitt zeigte sich ein hohe Strukturübereinstimmung der veröffentlichten Sequenzen mit der Referenzsequenz LC312715.1. Die berechnete Konsensussequenz zeigte relativ im Vergleich z.B. zu den SARS assoziierten Referenzen geringere Fehlerraten R1 = 8,60% und R2 = 8,83%.
Die Abbildung 6 zeigt deutliche Unterschiede zur Abbildung 13. Die Abdeckungsverteilung zeigt auch eher ein Wellenmuster mit relativ regelmäßigen Bereichen besonders hoher Abdeckung und unterschiedet sich daher deutlich von der Abdeckungsverteilung der zufälligen Referenz „rnd_uniform“. Die Verteilung der Leselängen (Abbildung 23b), vergleiche auch c)) unterscheidet sich ebenfalls deutlich zu den eher zufälligen Verteilungen und zeigt eine signifikante Anzahl mappbarer Lesungen mit einer Länge bis zu etwa 110 nt. Auch die durchschnittliche Leselänge liegt mit 51,84 nt höher, als z.B. bei „rnd_uniform“.
Interessant ist auch hier die Position der Primersequenzen in Bezug auf Bereiche mit hoher Nukleotidabdeckung im Vergleich zur mittleren Abdeckung. Es konnten insgesamt 46 der 52 Primersequenzen mit einer Fehlerrate von 38,00% der hier betrachteten Referenz zugeordnet werden. Die Abbildung 6 suggeriert, dass auch mit der Referenz LC312715.1 assoziierte kurze Sequenzlesungen im Rahmen der PCR-Bestätigung amplifiziert wurden und dies, obwohl die Primersequenzen nur mit einer relativ hohen Fehlerrate der Referenz zugeordnet werden können.
Kommen wir abschließend zu der Referenz KJ410048.1 (Masernvirus). Abbildung 10: Referenz KJ410048.1.
a) KJ410048.1_short_reads gemappt BBMap (M1; M2) = (37; 0,60).
b) KJ410048.1_primer gemappt mit BBMap
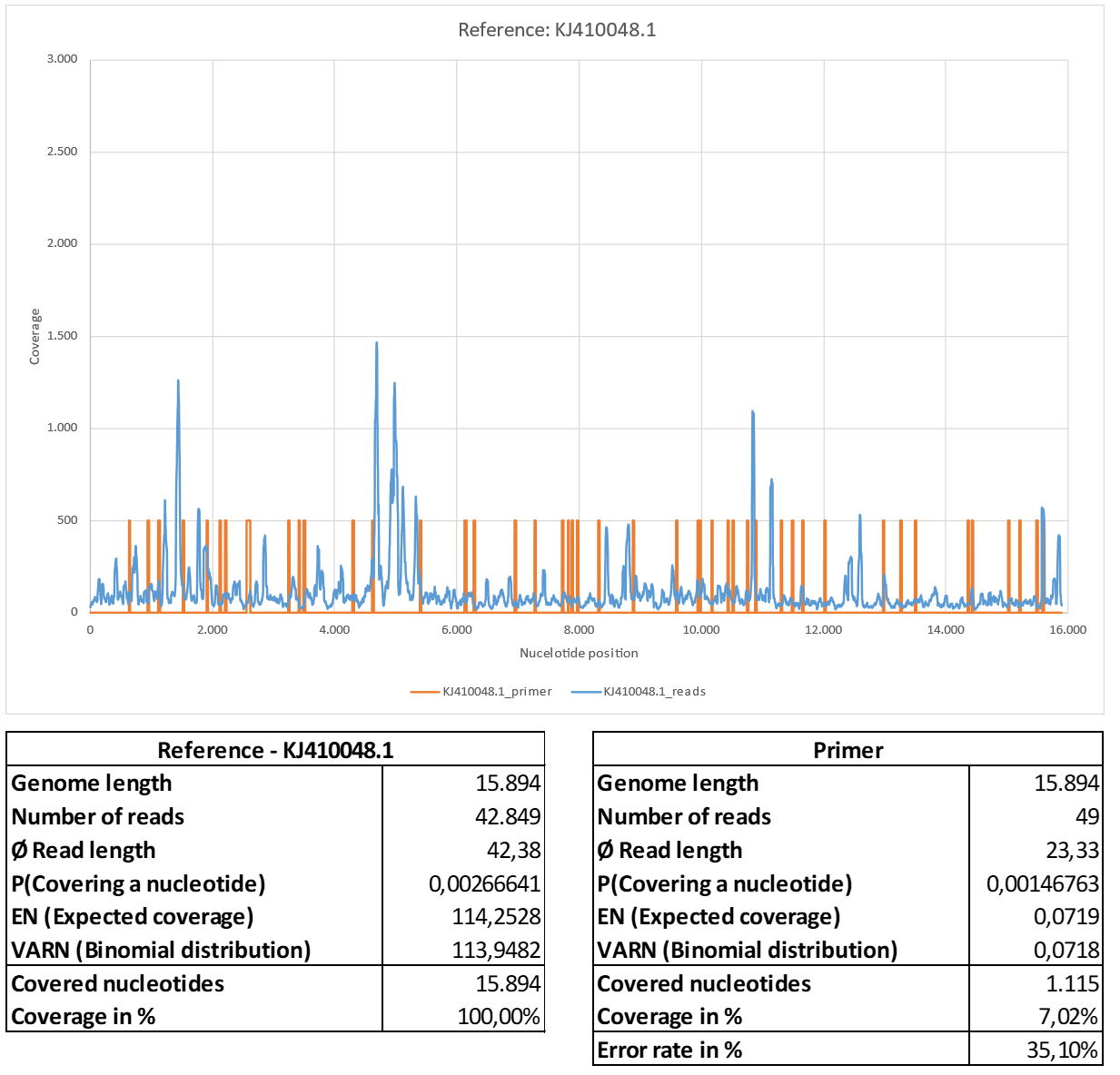
Die Abdeckungsverteilung weicht deutlich von der in Abbildung 6 ab und zeigt gewisse Ähnlichkeiten mit der Verteilung der assoziierten Sequenzlesungen bzgl. „rnd_uniform“, wobei die Schwankungen in Bereichen geringerer Abdeckung weniger stark ausgeprägt sind. Die Verteilung der Leselängen (Tabellen und Abbildungen: Abbildung 24d)) sowie die durchschnittliche Leselänge von 42,38 sind mit den Daten von „rnd_unifom“ vergleichbar und korrelieren ebenfalls mit relativ hohen Fehlerraten F1=28,70% und F2=28,79%.
Diskussion und Ausblick
Wir untersuchten mit einem einfachen bioinformatischen Protokoll die veröffentlichten Sequenzdaten (BioProject-Zugangsnummer PRJNA603194 in der Datenbank NCBI Sequence Read Archive (SRA)) zur Genomsequenz für SARS-CoV-2 (GenBank: MN908947.3). Die von uns verwendeten Methoden sind nicht spezifisch für SARS-CoV-2 und können ohne besondere Modifikationen auch auf andere Sequenzdaten angewendet werden.
Zunächst wiederholten wir mit den vorhandenen Sequenzdaten die Contigerstellung mit Megahit (v.1.2.9) und erhielten signifikant abweichende Resultate im Vergleich zu den Darstellungen in [1]. Wir konnten insbesondere das längste Contig mit 30.474 nt Länge, welches nach [1] nahezu das gesamte virale Genom umfasste und als Basis für das Primerdesign fungierte, nicht reproduzieren. Vielmehr zeigte das von uns generierte längste Contig (29.802 nt) eine nahezu vollständige Übereinstimmung mit der Referenz MN908947.3. Folglich kann es sich bei den veröffentlichten Sequenzdaten nicht um die ursprünglichen zur Contigerstellung verwendeten kurzen Lesungen handeln. Dies ist im Rahmen wissenschaftlicher Publikationen als überaus problematisch anzusehen, da so keine Überprüfung der veröffentlichten Ergebnisse mehr durchgeführt werden kann. Die Möglichkeit zur Überprüfung publizierter wissenschaftlicher Hypothesen ist das Wesen lebendiger Wissenschaft.
Im Widerspruch zu den Ausführungen in [1] haben wir Contigs hoher Abdeckungstiefe gefunden, welche wahrscheinlich mit (ribosomalen) Ribonukleinsäuren humanen Ursprungs assoziiert sind. Damit wurden nicht alle mit dem Menschen in Verbindung stehenden Nukleinsäuren während der Konstruktion von SARS-CoV-2 eliminiert. Weiter wurde kein Nachweis über die Existenz viraler Nukleinsäuren in der Patientenprobe erbracht und folglich besteht die Möglichkeit, dass zur Konstruktion der behaupteten viralen Sequenz MN908947.3 im bedeutenden Maße unbemerkt humane Nukleinsäurefragmente verwendet wurden. Diese Möglichkeit wäre durch Kontrollversuche auszuschließen.
In allen Publikationen zu den in dieser Studie analysierten Referenzgenome wurden die notwendigen Nachweise zur exakten Herkunft der zur Konstruktion verwenden Sequenzfragmente ebenfalls nicht erbracht und die notwendigen Kontrollversuche nicht publiziert.
Wir möchten an dieser Stelle noch erwähnen, dass möglicherweise bereits vielfach unbemerkt Kontrollexperimente durchgeführt wurden, die die Möglichkeit der Konstruktion von SARS-CoV-2 Genomen aus nicht infektiösen menschlichen Proben zeigen. So werden beispielsweise in [5] und [17] von Ganzgenomsequenzierungen aus Proben mit einem Ausgangs-Ct-Wert von über 35 berichtet. Dies könnte eine Widerlegung des viralen Modells für SARS-CoV-2 sein.
Die Analyse der Nukleotidabdeckungsverteilungen sowie die Längenverteilungen der mappbaren Sequenzlesungen für die jeweiligen Referenzsequenzen führt zu der Hypothese einer möglichen unbeabsichtigten Amplifikation von nicht mit SARS-CoV-2 assoziierten Sequenzlesungen. Weiter ist damit einhergehend die Möglichkeit der unbeabsichtigten Generierung von Sequenzen in Betracht zu ziehen, die in der Ausgangsprobe nicht enthalten waren, sondern nur durch die Amplifikationsbedingungen, wie z.B. die verwendeten Primersequenzen und die durchgeführten Zyklen, entstanden. Diese Möglichkeit erfordert daher die Durchführung geeigneter Kontrollversuche.
Neben dem Versuch der Wiederholung der in [1] publizierten Assemblierung mit den veröffentlichten Sequenzlesungen, haben wir ein einfaches Protokoll zur Analyse der inneren Struktur großer Datensätze kurzer Sequenzlesungen betrachtet. Mit den vorliegenden Sequenzdaten konnten wir für die Referenzgenome LC312715.1 (HIV) und NC_001653.2 (Hepatitis delta) Konsensussequenzen mit einer höheren Güte berechnen, als bei denen von uns betrachteten mit Coronaviren assoziierten Referenzsequenzen. Dies gilt insbesondere auch für bat-SL-CoVZC45 (GenBank: MG772933.1), welches zur Ursprungshypothese von SARS-CoV-2 führte.
Damit konnten wir unsere Hypothese erhärten, dass es sich bei den behaupteten viralen Genomsequenzen in dem Sinne um Fehldeutungen handelt, dass diese unbemerkt aus nicht viralen Nukleinsäurefragmenten konstruiert wurden bzw. werden. Insbesondere unterstreichen unsere Ergebnisse die dringliche Notwendigkeit zur Durchführung entsprechender Kontrollversuche. Für jede vermutete pathogene virale Genomsequenz bestünde ein naheliegendes Protokoll darin, jeweils aus entsprechenden unverdächtigen Proben unter Verwendung identischer Protokolle die Assemblierung der Genomsequenzen zu versuchen.
Wir beobachteten hohe Fehlerraten R1 und R2 bei den Referenzgenomen für Masern, Ebola oder Marburg, bei denen die zur Konstruktion verwendeten Nukleinsäurefragemente in Vero-Zellen propagiert wurden. Es bleibt bislang eine offene Frage, ob dies in den Nukleinsäurequellen selbst, oder an den verwendeten Amplifikationsbedingungen (z.B. Primersequenzen und Zyklenzahl) bzw. Sequenzierungsprotokollen (z.B. die verwendeten Polymerasen und reversen Transkriptasen) begründet liegt. Im Hinblick auf unsere Ergebnisse empfehlen wir neben der Veröffentlichung der endgültigen verwendeten Sequenzdaten stets auch solche, die nur durch die Amplifikation mit zufälligen Hexameren und moderaten Zyklenzahlen entstanden sind, um möglichst unverzerrte Daten zur Strukturanalyse bereitzustellen.
Material und Methoden
Abdeckungstiefe einer Referenzsequenz mit kurzen Sequenzlesungen
Es bezeichne G die Länge der Referenzsequenz, ØL die durchschnittliche Leselänge, n die Anzahl der kurzen Sequenzlesungen sowie N die zufällige, durchschnittliche Abdeckungstiefe der Referenzsequenz mit den kurzen Sequenzlesungen. Dann gilt
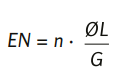
Der Ausdruck kann als Wahrscheinlichkeit der Abdeckung eines Nukleotids innerhalb der Referenzsequenz mit einer kurzen Sequenzlesung angesehen werden.
Generierung zufälliger Referenzsequenzen
Der nachfolgende Satz ermöglicht die Simulation einer Zufallsgröße X mit kumulativer Verteilungsfunktion F.
Satz (Inversionsprinzip) [28].
Es sei U eine auf dem Intervall (0,1) gleichverteilte Zufallsgröße. Weiter seien X eine Zufallsgröße mit kumulativer Verteilungsfunktion F, sowie
F-1 (y) := inf {x ∈ ℝ|F(x)≥y}.
Dann gilt
F-1 (U) ~ X.
Es seien Ui ,i=1,…,29.903 unabhängig identische gleichverteilte Zufallsgrößen auf dem Intervall (0,1). Mit pnt,nt ∈{A,T,C,G} bezeichne die Wahrscheinlichkeit für das Nukleotid nt. Dann ergibt sich das Nukleotid Ni ,i=1,…,29.903 der zufällig generierten Referenzsequenz via
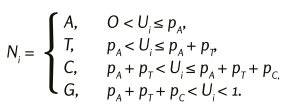
Bei der Referenzsequenz „rnd_unifom“ wurde die Gleichverteilung auf der Menge {A,T,C,G} verwendet. Zur Simulation der zufälligen Referenzsequenz „rnd_wuhan“ wurde als Nukleotidverteilung das relative Auftreten der Nukleotide A, T, C und G in der Genomsequenz für SARS-CoV-2 (GenBank: MN908947.3) gewählt. Bei der Konstruktion der randomisierten Referenzsequenzen „rnd_wh_mk_1“ und „rnd_wh_mk_2“ wurde jeweils die bedingte Wahrscheinlichkeit, bedingt auf das letzte bzw. auf die letzten beiden Nukleotide gemäß den entsprechenden empirischen Häufigkeiten in der Sequenz für SARS-CoV-2 (GenBank: MN908947.3) gewählt.
Stochastische Simulation zufälliger Abdeckungen einer Referenzsequenz
Die kumulative Verteilungsfunktion der Exponentialverteilung mit Parameter λ lautet [28],
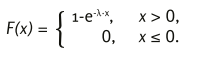
Es sei X eine Zufallsgröße mit Verteilungsfunktion F. Dann gilt

Bioinformatische Methoden (Strukturanalyse)
1. Mapping mit BBMap
bbmap.sh ref=$reference.fasta
mapPacBio.sh in=SRR10971381_1.fastq in2=SRR10971381_2.fastq outm=mapped.sam vslow k=8 maxindel=0 minratio=0.1
2. Selektion der gemappten Sequenzen in Abhängigkeit von M1 und M2 mit BBMap (reformat.sh)
reformat.sh in=mapped.sam out=sample_selection. sam minlength=$M1 (maxlength=100) idfilter=$M2 ow=t
3. Berechnung der Konsensussequenz
3.1. Vorbereitung mit Samtools
samtools view -b sample_selection.sam > sample.bam samtools sort sample.bam -o sample_sort_reads.bam samtools index sample_sort_reads.bam
3.2 Rohe Konsensussequenz
samtools mpileup -uf mapping/$reference.fasta sample_sort_reads.bam | bcftools call -c | vcfutils. pl vcf2fq > SAMPLE_cns.fastq
3.3 Finale Konsensussequenz (min. Q20)
seqtk seq -aQ64 -q20 -n N sample_cns.fastq > sample_cns.fasta
4. Mapping der Konsensussequenz an die Referenzsequenz mit BWA
bwa index $reference.fasta bwa mem $reference.fasta sample_cns.fasta > sample_cns.sam
5. Begutachtung mit Tablet
Die Begutachtung erfolgte mit der Visualisierungssoftware Tablet für die Visualisierung von Sequenzdaten.
Referenzen / Quellenverweis
[1] Fan Wu u. a. A new coronavirus associated with human respiratory disease in China. In: Nature 580.7803 (2020). DOI: 10.1038/s41586-020-2202-3.
[2] Na Zhu u. a. A Novel Coronavirus from Pati ents with Pneumonia in China, 2019. In: New England Journal of Medicine 382.8 (2020), S. 727-733. DOI:10.1056/nejmoa2001017.
[3] Divinlal Harilal u. a. SARS-CoV-2 Whole Genome Amplication and Sequencing for Effective Population-Based Surveillance and Control of Viral Transmission. In: Clinical Chemistry 66.11 (2020), S. 1450-1458. DOI: 10.1093/clinchem/hvaa187.
[4] Jalees A. Nasir u. a. A Comparison of Whole Genome Sequencing of SARSCoV-2 Using Amplicon-Based Sequencing, Random Hexamers, and Bait Capture. In: Viruses 12.8 (2020), S. 895. DOI: 10.3390/v12080895.
[5] Clinton R. Paden u. a. Rapid, sensitive, full-genome sequencing of severe acute respiratory syndrome coronavirus 2. In: Emerging Infectious Diseases 26.10 (2020), S. 2401-2405. DOI: 10.3201/ eid2610.201800.
[6] Sureshnee Pillay u. a. Whole Genome Sequencing of SARS-CoV-2: Adapting Illumina Protocols for Quick and Accurate Outbreak Investigation during a Pandemic. In: Genes 11.8 (2020), S. 949. DOI: 10.3390/genes11080949.
[7] Dan Hu u. a. Genomic characterization and infectivity of a novel SARS-like coronavirus in Chinese bats. In: Emerging Microbes & Infections 7.1 (2018), S. 1-10. DOI: 10.1038/s41426-018-0155-5.
[8] Davaalkham Jagdagsuren u. a. The second molecular epidemiological study of HIV infection in Mongolia between 2010 and 2016. In: Plos One 12.12 (2017). DOI: 10.1371/journal.pone.0189605.
[9] J. A. Saldanha, H. C. Thomas und J. P. Monjardino. Cloning and sequencing of RNA of hepatitis delta virus isolated from human serum. In: Journal of General Virology 71.7 (1990), S. 1603-1606. DOI: 10.1099/0022-1317-71-7-1603.
[10] Jernej Mlakar u. a. Zika Virus Associated with Microcephaly. In: New England Journal of Medicine 374.10 (2016), S. 951-958. DOI: 10.1056 /nejmoa1600651.
[11] Christopher L. Parks u. a. Comparison of Predicted Amino Acid Sequences of Measles Virus Strains in the Edmonston Vaccine Lineage. In: Journal of Virology 75.2 (2001), S. 910-920. DOI: 10.1128/ jvi.75.2.910-920.2001.
[12] Konstantin M. J. Sparrer u. a. Complete Genome Sequence of a Wild-Type Measles Virus Isolated during the Spring 2013 Epidemic in Germany. In: Genome Announcements 2.2 (2014). DOI: 10.1128/ genomea.00157-14.
[13] Paul A. Rota u. a. Characterization of a Novel Coronavirus Associated with Severe Acute Respiratory Syndrome. In: Science 300.5624 (2003), S. 1394-1399. DOI: 10.1126/science.1085952.
[14] Runtao He u. a. Analysis of multimerization of the SARS coronavirus nucleocapsid protein. In: Biochemical and Biophysical Research Communications 316.2 (2004), S. 476-483. DOI: 10.1016/j. bbrc.2004.02.074.
[15] Tracey Goldstein u. a. The discovery of Bombali virus adds further support for bats as hosts of ebolaviruses. In: Nature Microbiology 3.10 (2018), S. 1084- 1089. DOI: 10.1038/s41564-018-0227-2.
[16] Jonathan S. Towner u. a. Marburgvirus Genomics and Association with a Large Hemorrhagic Fever Outbreak in Angola. In: Journal of Virology 80.13 (2006), S. 6497-6516. DOI: 10.1128/jvi.00069-06.
[17] Annika Brinkmann u. a. Amplicov: Rapid whole-genome sequencing using multiplex PCR amplication and real-time Oxford Nanopore minion sequencing enables rapid variant identication of SARS-COV-2. In: Frontiers in Microbiology 12 (2021). DOI: 10.3389/fmicb.2021.651151.
[18] SARS-COV-2. url: https://artic.network/ncov-2019.
[19] Ncbi. ncbi/sra-tools: SRA Tools. URL: https://github.com/ncbi/sra-tools.
[20a] Dinghua Li u. a. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. In: Bioinformatics 31.10 (2015), S. 1674-1676. DOI: 10.1093/bioinformatics/btv033.
[20b] Voutcn. voutcn/megahit: Ultra-fast and memory-ecient (meta-)genome assembler. URL: https://github.com/voutcn/megahit.
[21a] Shifu Chen u. a. fastp: an ultra-fast all-inone FASTQ preprocessor. In: Bioinformatics 34.17 (2018), S. i884-i890. DOI: 10.1093/bioinformatics/ bty560.^
[21b] OpenGene. OpenGene/fastp: An ultra-fast all-in-one FASTQ preprocessor (QC/adapters/trimming/ltering/splitting/merging...)
URL: https://github.com/OpenGene/fastp.
[22a] Ben Langmead u. a. Scaling read aligners to hundreds of threads on generalpurpose processors. In: Bioinformatics 35.3 (2018), S. 421-432. DOI: 10. 1093/bioinformatics/bty648.
[22b] Ben Langmead. BenLangmead/bowtie2: A fast and sensitive gapped read aligner. URL: https://github.com/BenLangmead/bowtie2.
[23a] Brian Bushnell. BBMap: A Fast, Accurate, Splice-Aware Aligner. In: (March 2014). URL: https://www.osti.gov/biblio/1241166.
[23b] BBMap. url: https://sourceforge.net/projects/bbmap/.
[24a] Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. In: (May 2013). URL: https://arxiv.org/abs/1303.3997.
[24b] lh3. lh3/bwa: Burrow-Wheeler Aligner for short-read alignment (see mini-map2 for long-read alignment). URL: https://github.com/lh3/bwa.
[25a] H. Li u. a. The Sequence Alignment/Map format and SAMtools. In: Bioinformatics 25.16 (2009), S. 2078-2079. DOI: 10.1093/bioinformatics/btp352.
[25b] Samtools. url: http://www.htslib.org/.
[25c] Petr Danecek u. a. Twelve years of SAMtools and BCFtools. In: GigaScience 10.2 (2021). DOI: 10.1093/gigascience/giab008.
[25d] P. Danecek u. a. The variant call format and VCFtools“. In: Bioinformatics 27.15 (2011), S. 2156- 2158. DOI: 10.1093/bioinformatics/btr330. [26] Tablet. URL: https://ics.hutton.ac.uk/tablet/.
[27a] Wei Shen u. a. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. In: Plos One 11.10 (2016). DOI: 10.1371/journal.pone.0163962.
[27b] lh3. lh3/seqtk: Toolkit for processing sequences in FASTA/Q formats.
URL: https://github.com/lh3/seqtk.
[28] Albrecht Irle. Wahrscheinlichkeitstheorie und Statistik: Grundlagen - Resultate - Anwendungen. Teubner, 2010.
Unsere Buchbandreihe "Die Zeitzeugen" ❗️

➖ Was wir in über 30 Jahren intensivster Recherche, eigenständig finanzierten Kontrollexperimenten, gewonnenen Gerichtsverfahren, persönlichem Austausch, sowie Schriftverkehr mit den weltweit führenden Virologen und Institutionen herausgearbeitet haben, übersteigt die Vorstellungskraft der meisten Menschen.
➖ Jedes Buch beinhaltet eigenständiges Wissen, welches jeden Staat, Politiker, Wissenschaftler und Bürger in die Handlung zwingt.
➖ Unser Versprechen: Wir BEWEISEN, dass ALLE Existenzbehauptungen krankmachender Viren, Pandemien, die Ansteckungstheorie sowie Impfstoffe auf einem Irrtum beruhen und eine Gefahr für Leib und Leben darstellen.
Fragen? Unser Ansprechpartner auf Telegram steht ihnen zur Verfügung !
Klick 👉 @NotIsolate
Mit dem Kauf eines der Bücher "Die Zeitzeugen" unterstützen Sie gleichzeitig unsere Arbeit und weitere Buch-Bände. Wir Danken Ihnen sehr ❤️🙏🏻
👇 J e t z t B e s t e l l e n 👇
📚 --> Die Zeitzeugen Band 1.0 | [Promo Video]
📚 --> Die Zeitzeugen Band 1.1 | [Promo Video]
Telegram-Hauptkanal: https://t.me/Corona_Fakten
Ansprechpartner auf Telegram für den premium Access:
Benutzername: @NotIsolate
Folgend eine Liste unserer wichtigsten Artikel: