In Autosomal Dominant Inheritance Aspx Openfile

⚡ ALL INFORMATION CLICK HERE 👈🏻👈🏻👈🏻
In Autosomal Dominant Inheritance Aspx Openfile
Health Encyclopedia Home
Tests & Procedures
Interactive Encyclopedia Tools
Healthy Living
Your Family
Drug Reference
Herbs, Vitamins & Supplements
Prevention Planner
Tests & Procedures
Interactive Encyclopedia Tools
Healthy Living
Your Family
Drug Reference
Herbs, Vitamins & Supplements
Prevention Planner
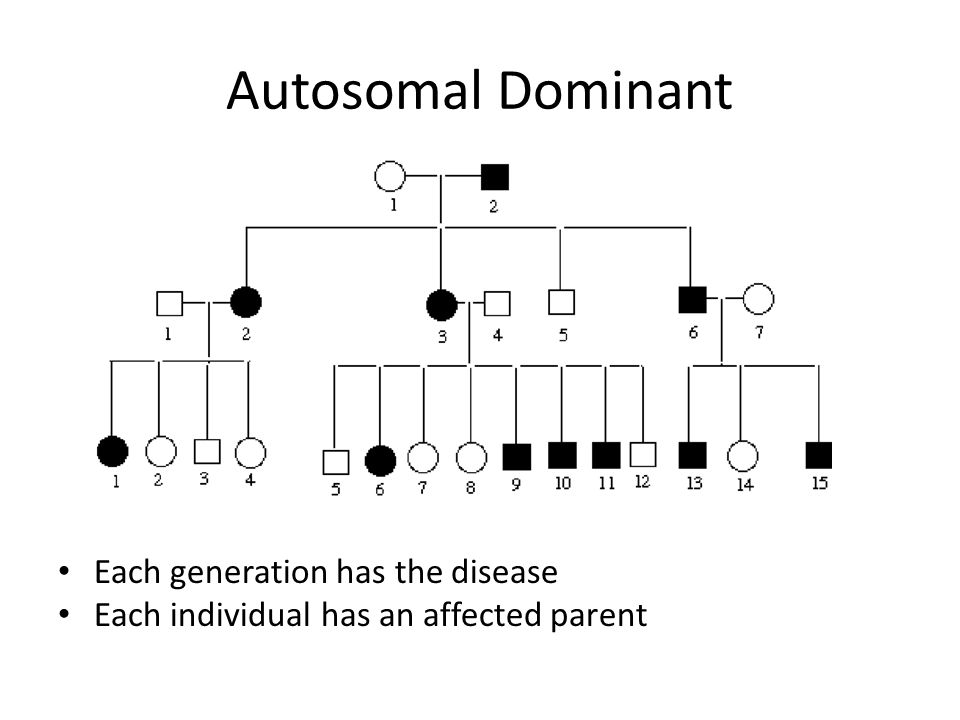
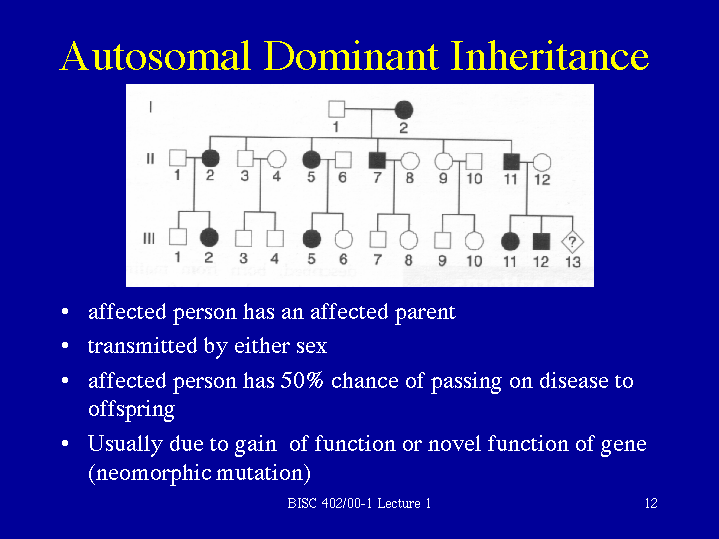
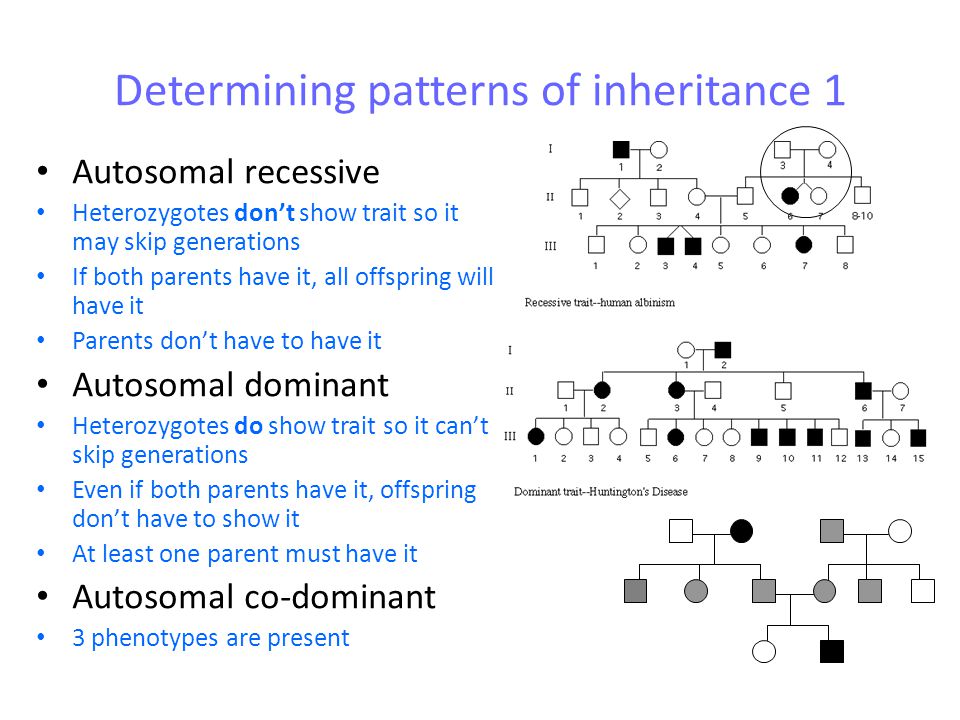
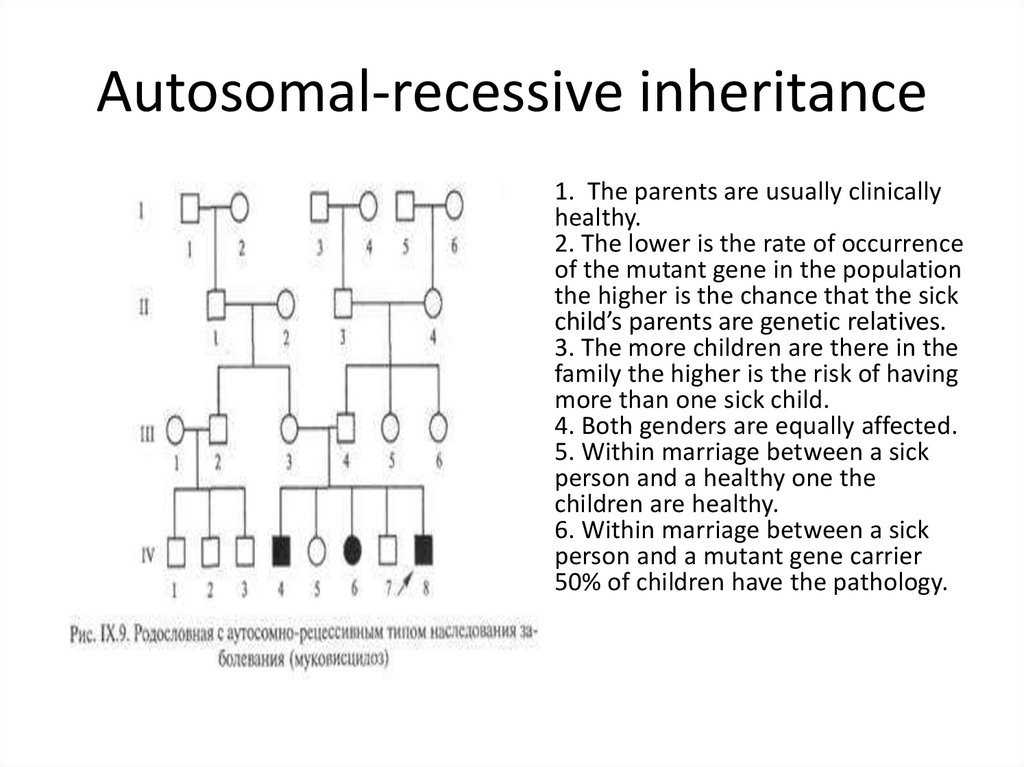
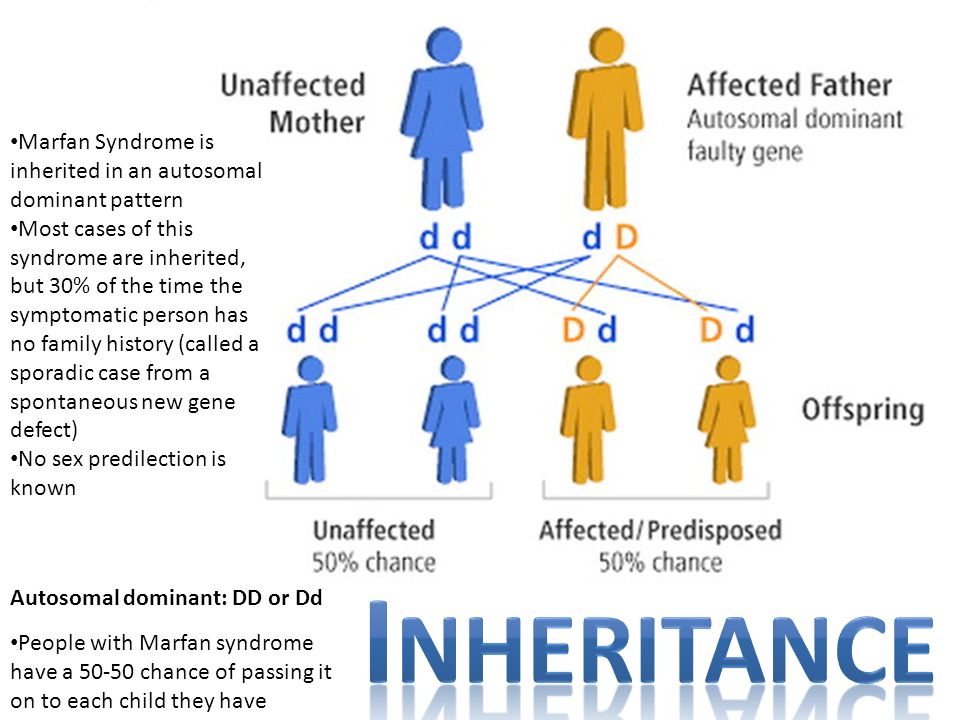
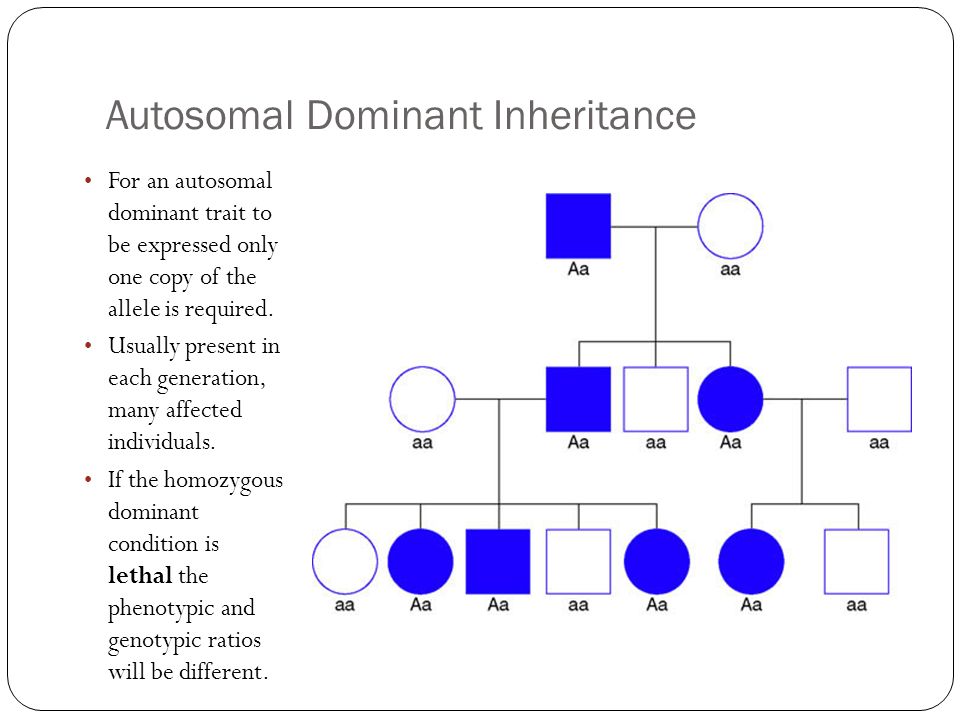
Genes are the blueprints for making proteins. Our bodies need proteins to develop
and work properly. Most genes come in pairs. One is inherited from the mother and
the other from the father. Genes inherited from our biological parents are expressed
in specific ways. One of these basic patterns is called autosomal dominant inheritance.
Sex chromosomes, which determine male or female gender
Autosomes, which are all of the other chromosomes (chromosome pairs 1 through 22)
or nonsex chromosomes
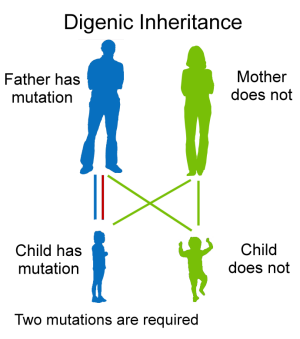
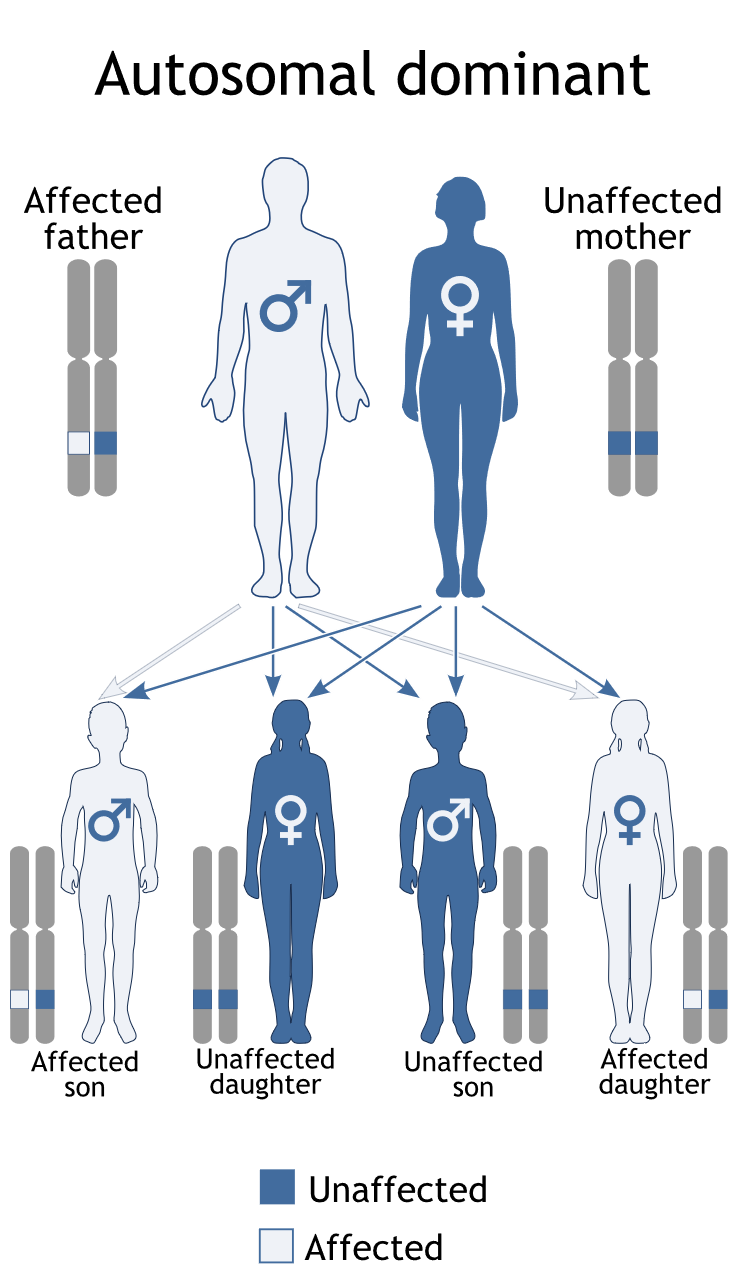
Autosomal inheritance of a gene means that the gene is located on one of the autosomes.
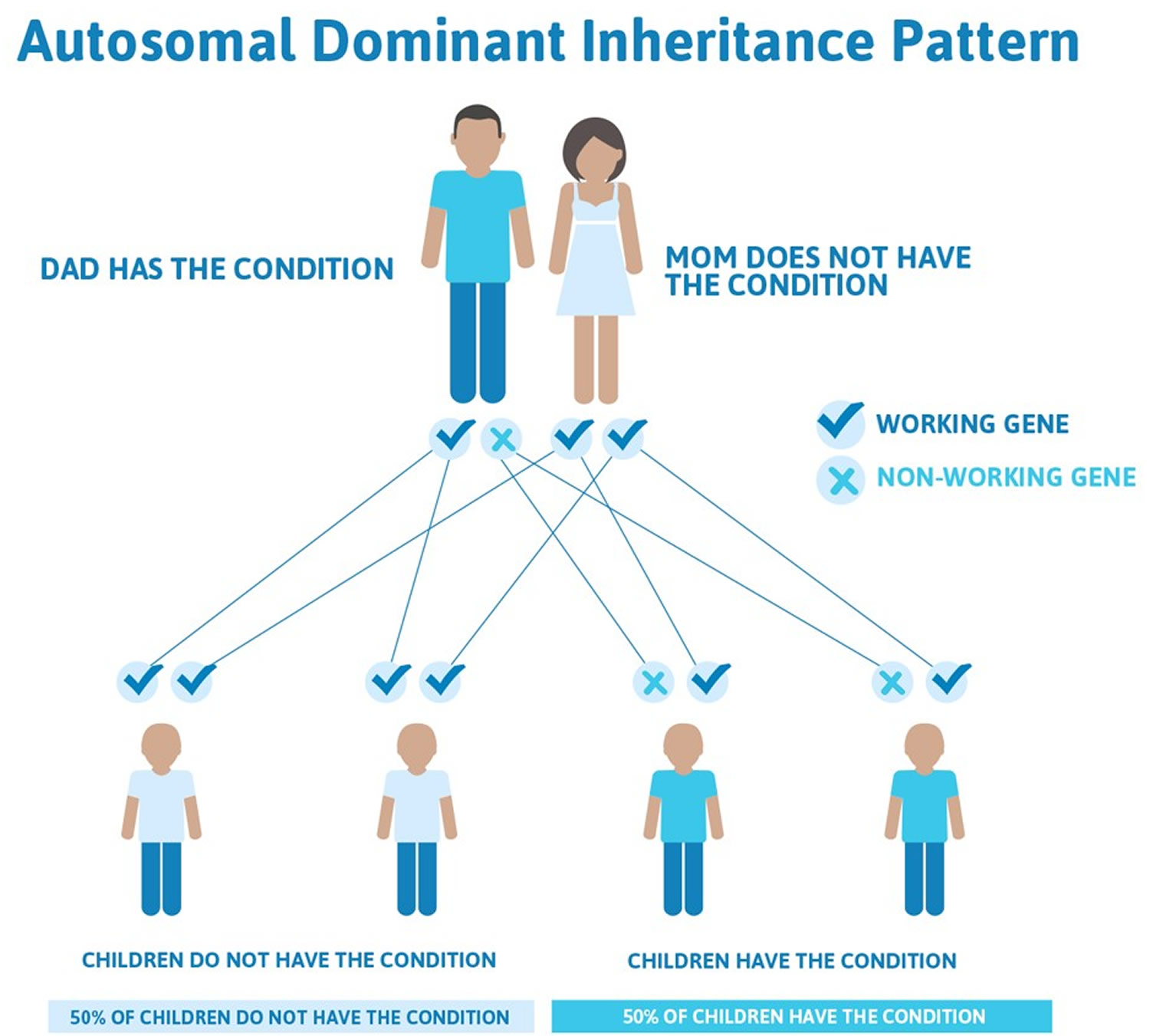
This means that males and females are equally likely to inherit the gene. "Dominant"
means that a single copy of the gene can cause a particular trait, such as brown eyes
instead of blue eyes. When a parent has a dominant gene, there is at least a 50% chance
that any child they have will also have the trait.
There are 4 possible combinations in the children (see figure). These combinations
are possible every time a pregnancy occurs between these 2 individuals. The gender
of the children (whether they are sons or daughters) does not matter. The chance is
50/50 for them to inherit the autosomal genes.
A characteristic of some dominant genes is that they can have variable expression. This
means that some people have milder or more intense characteristics than others. Another
important characteristic of dominant genes is that, in some cases, they can have reduced
penetrance. This means that sometimes a person can have a dominant gene copy but not
show any signs of the gene. The concept of reduced penetrance is particularly important
in the case of autosomal dominant cancer susceptibility genes. If a person has inherited
a cancer susceptibility gene, it does not mean they will automatically develop cancer.
It simply means that the person has inherited a mutation in a gene that gives them
a higher chance to develop cancer than someone without the mutation.
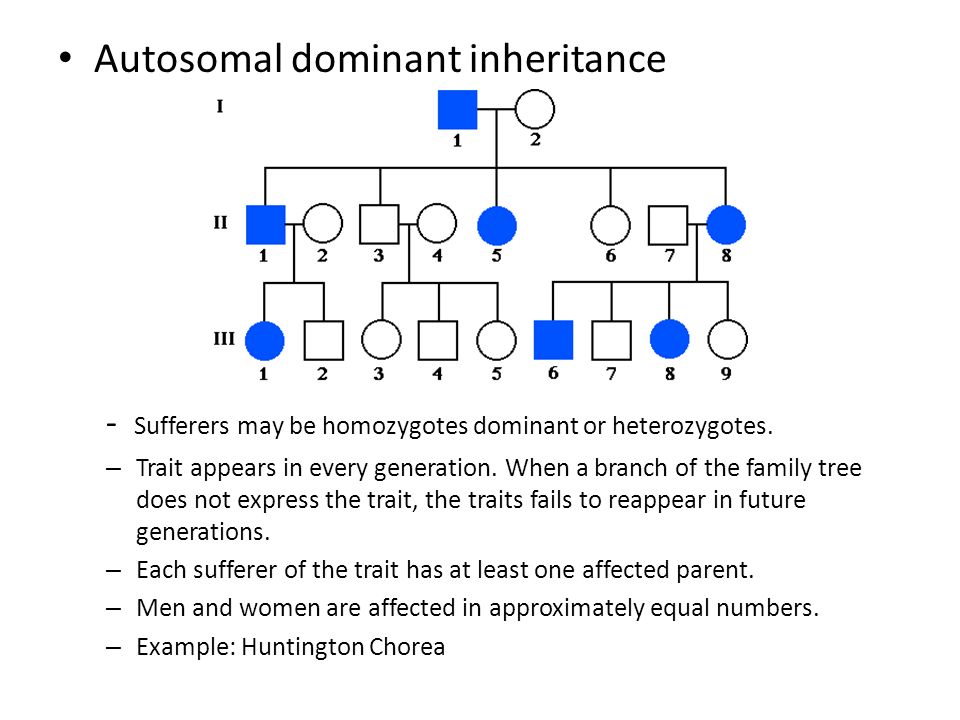
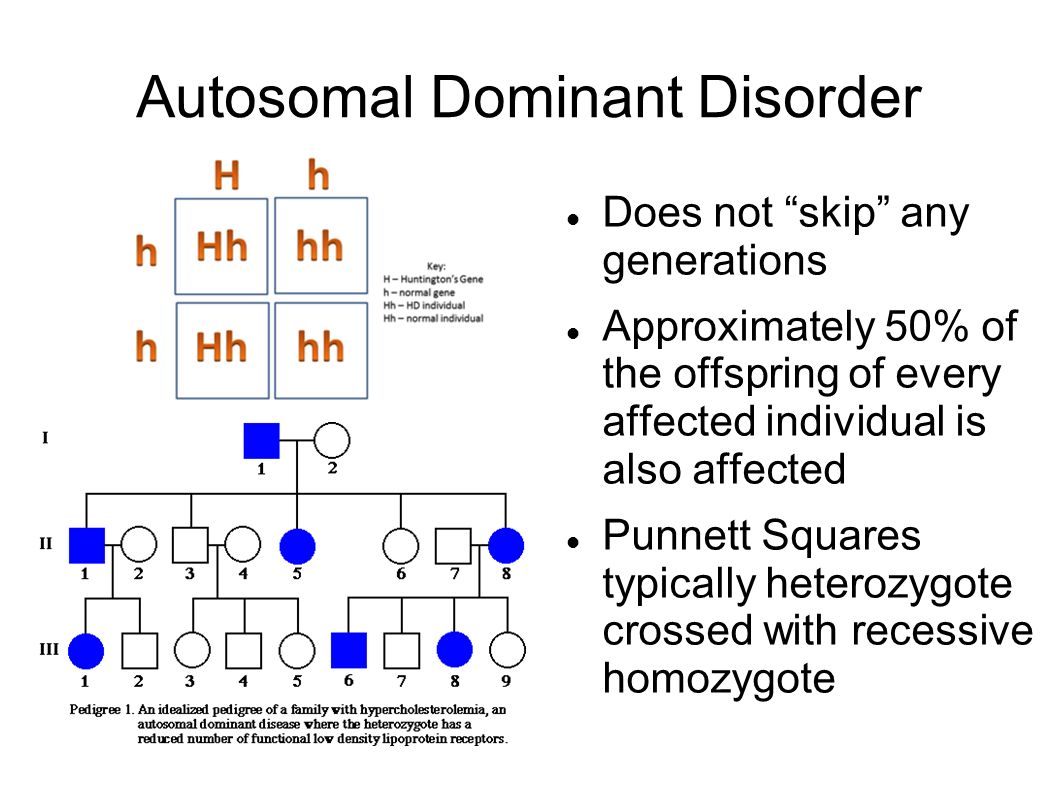
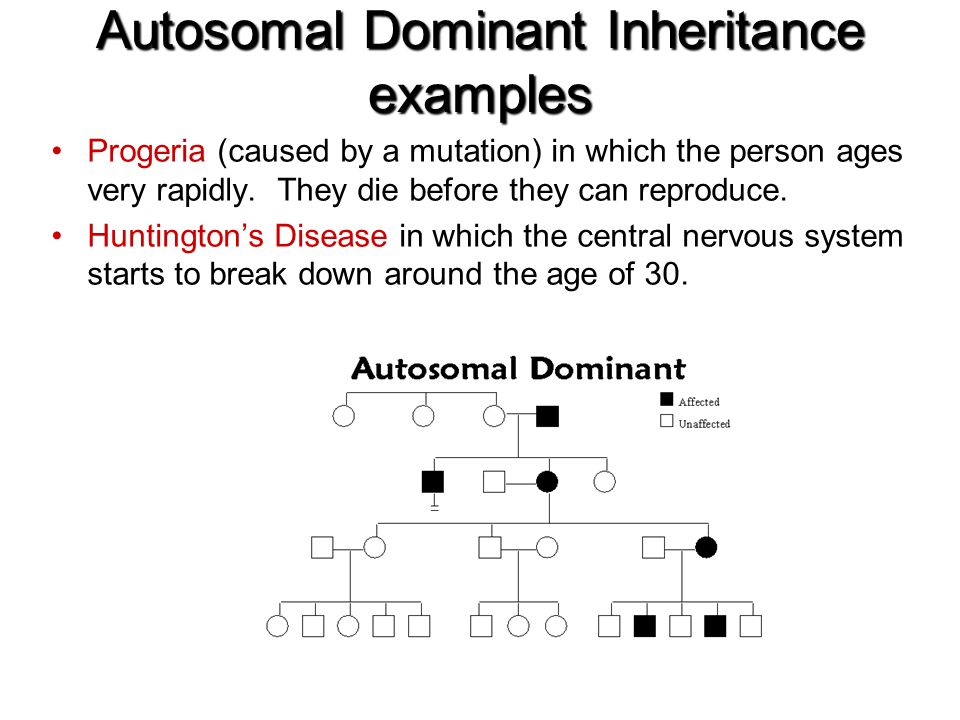
Examples of conditions involving autosomal dominant inheritance are:
©2021 University of Rochester Medical Center Rochester, NY
In Autosomal Dominant Inheritance Aspx Openfile – Telegraph
Autosomal Dominant Inheritance - Health Encyclopedia...
(PDF) Autosomal dominant inheritance of Weaver syndrom
Autosomal Dominant Inheritance Calculator
Figure 1: Autosomal Dominant Inheritance
Most report of Weaver syndrome have been sporadic cases and the genetic basis of the syndrome is uncertain. This report of an affected father and daughter provides evidence for autosomal dominant inheritance.
Appearance of case 1 at 3 years of age with her father (case 2).
Appearance of case 2 (boy on the right of the photograph) during childhood.
Appearance of the finger nails of case 2.
Content may be subject to copyright.
Content may be subject to copyright.
... In addition, there is a hypothesized increased risk for tumorgenesis but further investigation is necessary given the likely large reporting bias and potential impact this association has on development of specific screening protocols [Lapunzina, 2005;Basel-Vanagaite, 2010]. The majority of cases are sporadic, however, familial cases with autosomal dominant inheritance have been described [Fryer et al., 1997; Proud et al., 1998]. Pathogenic variants in EZH2, a histone methyltransferase, have previously been identified as a cause of Weaver syndrome [Gibson et al., 2011;Tatton-Brown et al., 2011]. ...
Weaver syndrome is a rare condition characterized by overgrowth, macrocephaly, accelerated osseous maturation, variable intellectual disability, and characteristic facial features. Pathogenic variants in EZH2, a histone methyltransferase, have previously been identified as a cause of Weaver syndrome. However, the underlying molecular cause in many patients remains unknown. We report a patient with a clinical diagnosis of Weaver syndrome whose exome was initially non-diagnostic. Reports in the medical literature of EED associated overgrowth prompted re-analysis of the patient's original exome data. The patient was found to have a likely pathogenic variant in EED. These findings support that Weaver syndrome is a disorder with locus heterogeneity and can be due to pathogenic variants in either EZH2 or EED. This case highlights the utility of exome sequencing as a clinical diagnostic tool for novel gene discovery as well as the importance of re-examination of exome data as new information about gene-disease associations becomes available. © 2016 Wiley Periodicals, Inc.
... Key words: EZH2; Weaver syndrome; histone methyl transferases INTRODUCTION Weaver et al. [1974] described two boys with accelerated osseous maturation, unusual facies, and camptodactyly and proposed that this constellation of phenotypic features constituted a novel clinical entity. Subsequent to this initial description over 50 individuals with similar and additional clinical features were reported and the condition became eponymously known as Weaver syndrome (OMIM 277590) [Majewski et al., 1981;Meinecke et al., 1983;Roussounis and Crawford, 1983;Tsukahara et al., 1984;Farrell and Hughes, 1985;Ardinger et al., 1986;Thompson et al., 1987;Greenberg et al., 1989;Kondo et al., 1990Kondo et al., , 1991Muhonen and Menezes, 1990;Ramos-Arroyo et al., 1991;Cole et al., 1992;Dumic et al., 1993;Scarano et al., 1996; Fryer et al., 1997; Proud et al., 1998;Derry et al., 1999;Freeman et al., 1999;Sarigul et al., 1999;Kelly et al., 2000;Ozkan and Bereket, 2000;Huffman et al., 2001;Crawford and Rohan, 2005;Coulter et al., 2008;Iatrou et al., 2008;Bansal and Bansal, 2009;Basel-Vanagaite, 2010;Mikalef et al., 2010]. However, because of the subtlety of the Weaver syndrome phenotype and the overlap with other overgrowth syndromes, particularly Sotos syndrome, the clinical diagnosis can be challenging, even for the experienced dysmorphologist. ...
Weaver syndrome, first described in 1974, is characterized by tall stature, a typical facial appearance, and variable intellectual disability. In 2011, mutations in the histone methyltransferase, EZH2, were shown to cause Weaver syndrome. To date, we have identified 48 individuals with EZH2 mutations. The mutations were primarily missense mutations occurring throughout the gene, with some clustering in the SET domain (12/48). Truncating mutations were uncommon (4/48) and only identified in the final exon, after the SET domain. Through analyses of clinical data and facial photographs of EZH2 mutation-positive individuals, we have shown that the facial features can be subtle and the clinical diagnosis of Weaver syndrome is thus challenging, especially in older individuals. However, tall stature is very common, reported in >90% of affected individuals. Intellectual disability is also common, present in ∼80%, but is highly variable and frequently mild. Additional clinical features which may help in stratifying individuals to EZH2 mutation testing include camptodactyly, soft, doughy skin, umbilical hernia, and a low, hoarse cry. Considerable phenotypic overlap between Sotos and Weaver syndromes is also evident. The identification of an EZH2 mutation can therefore provide an objective means of confirming a subtle presentation of Weaver syndrome and/or distinguishing Weaver and Sotos syndromes. As mutation testing becomes increasingly accessible and larger numbers of EZH2 mutation-positive individuals are identified, knowledge of the clinical spectrum and prognostic implications of EZH2 mutations should improve.
Genetisch bedingter Fehlbildungskomplex auf der Grundlage einer Genmutation.
Advances in molecular biology, cell biology, and several other areas of science have changed the way we understand the mechanisms in which microbial pathogens interact with their hosts. This trend is set to continue with the advent of microbial genome sequencing, in vivo gene expression analysis, and other related techniques. The availability of these techniques and advances in other areas such as protein expression and crystallography has allowed the understanding of host pathogen interaction at the molecular and even atomic level. However, despite these powerful approaches the basic concept advanced many years ago by Smith that pathogenicity or virulence is a multifactorial property that consists of five basic steps is still valid today.1 The molecular basis of virulence can still be considered under these five headings: (1) attachment to the host (via mucous membranes); (2) entry into the host (usually); (3) multiplication within the host; (4) Interference with host defence systems; (5) damage to the host.
These five stages are not mutually exclusive. The factors produced by pathogens that mediate these steps are termed the determinants of microbial pathogenicity. The molecular basis of these steps will be considered and specific examples will be given to demonstrate the basic principles. Finally it should be emphasised that expression of determinants of pathogenicity is usually regulated and systems exist for environmental sensing and quorum sensing to allow appropriate expression of virulence factors.
Attachment of bacteria to host cells is mediated by adhesins, which have been identified for many bacterial species.2-5Most adhesins are proteins which usually bind to carbohydrate receptors on the host cell surface. Perhaps the most studied adhesins are the fimbrial adhesins of Escherichia coli . These can be divided into two families. The K88, K99, CFA/I, and CFA/II adhesins mediate attachment to gut epithelium while type 1, P, …
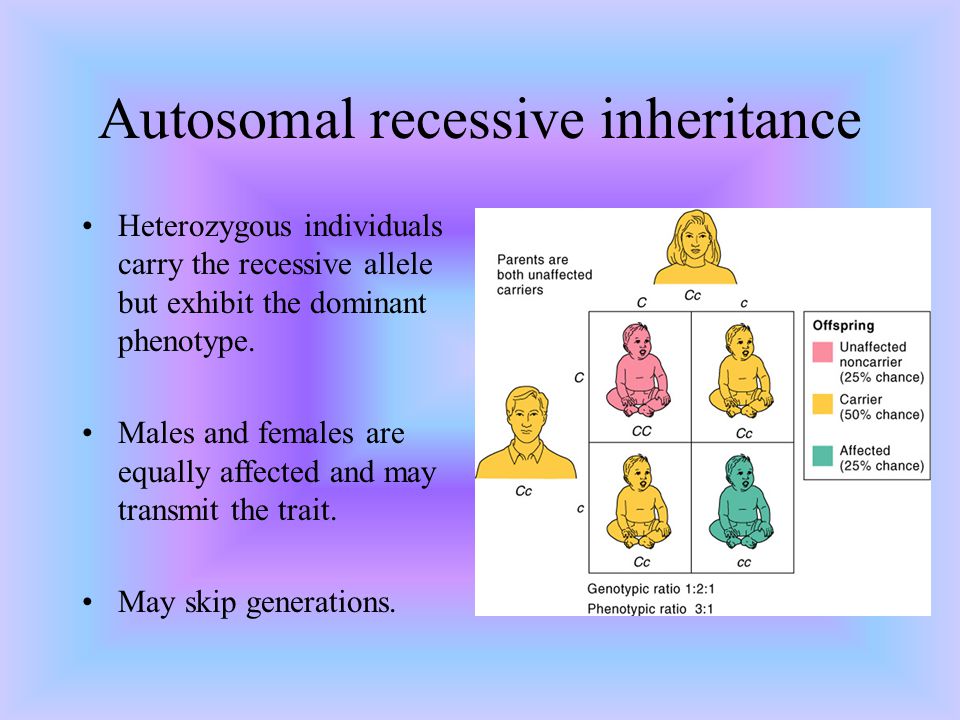
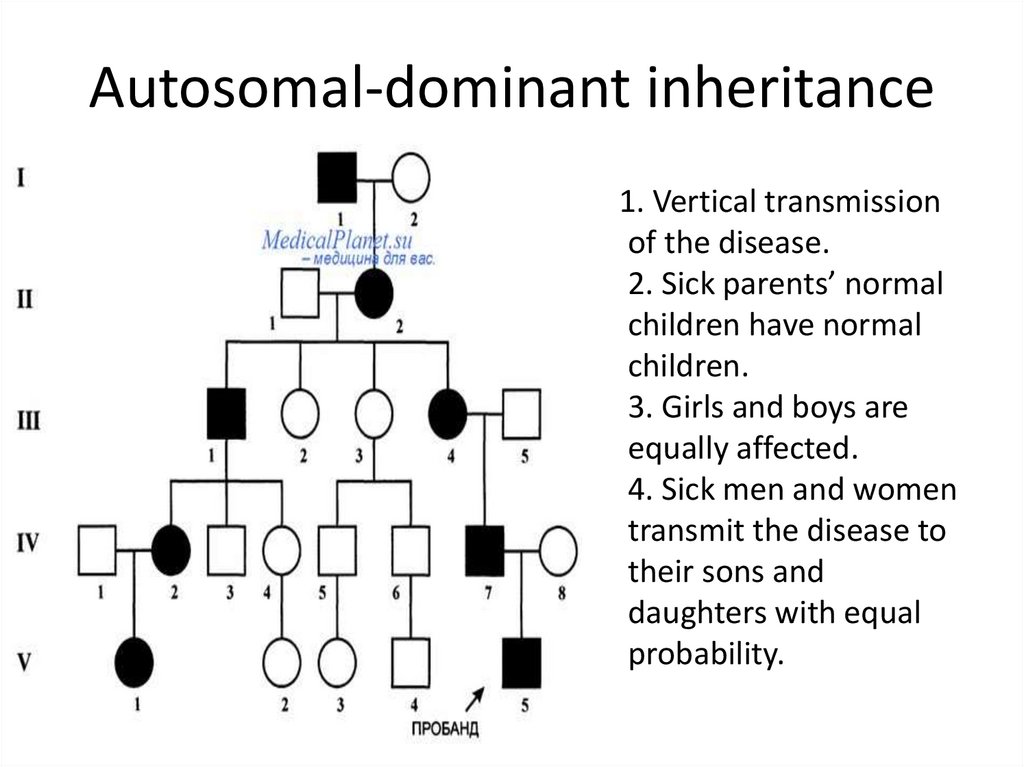
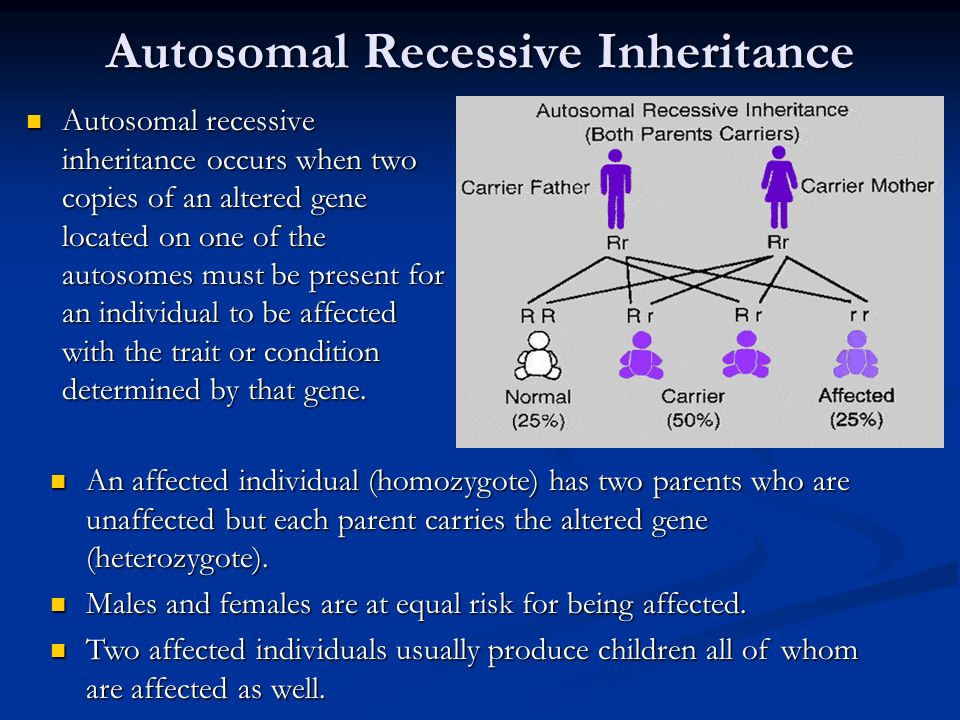
Benign recurrent intrahepatic cholestasis (BRIC) is an autosomal recessive liver disease characterized by multiple episodes of cholestasis without progression to chronic liver disease. On the basis of recent evidence of locus heterogeneity, we studied 19 subjects (7 affected members) of a BRIC family. Male-to-male transmission and the presence of affected females suggested autosomal dominant inheritance. Blood samples were collected after informed consent. Subjects were genotyped by using markers mapping to 18q and 2q24 region, respectively, where the genes FIC1 and FIC2 have been mapped. Segregation of haplotypes excluded the two regions in our family. These findings suggest further genetic heterogeneity of the origin of BRIC. Am. J. Med. Genet. 95:450–453, 2000. © 2000 Wiley-Liss, Inc.
We report on an infant with Weaver syndrome, neoplasia and cardiovascular anomalies. Stage 4S neuroblastoma underwent spontaneous resolution. Three neoplasms have been reported in Weaver syndrome: another stage 4S neuroblastoma [Muhonen and Menezes, 1990: J Pediatr 116:596–599], an ovarian endodermal sinus tumor [Derry et al., 1999: J Med Genet 36:725–728], and a sacrococcygeal teratoma [Kelly et al., 2000: Am J Med Genet 95:492–495]. No case was associated with cardiovascular anomalies. Our patient had VSD and PDA, and although several other patients with Weaver syndrome have had cardiovascular anomalies, they were shown not to have neoplasia. © 2001 Wiley-Liss. Inc.
Weaver syndrome is an autosomal dominant disorder comprising accelerated growth rate and rapidly advancing skeletal maturation. Previous reports suggest that the phenotype in adults may be sufficiently subtle to make diagnosis difficult. Half brothers with classical childhood findings of Weaver syndrome and their father with minimal clinical findings showed cervical spine anomalies that likely represent a consistent radiographic finding in this disorder. One of the children represents the third occurrence of neoplasia in Weaver syndrome. Am. J. Med. Genet. 95:492–495, 2000. © 2000 Wiley-Liss, Inc.
January 1969 · Rivista di Patologia Nervosa e Mentale
July 1985 · Australian Dental Journal
— Orthodontic diagnosis and treatment planning should include an assessment of a child's skeletal development. A case history illustrates the futility of relying entirely on chronological age.
November 2009 · Developmental Dynamics
Robinow syndrome is a skeletal dysplasia with both autosomal dominant and autosomal recessive inheritance patterns. It is characterized by short stature, limb shortening, genital hypoplasia, and craniofacial abnormalities. The etiology of dominant Robinow syndrome is unknown; however, the phenotypically more severe autosomal recessive form of Robinow syndrome has been associated with mutations in ... [Show full abstract] the orphan tyrosine kinase receptor, ROR2, which has recently been identified as a putative WNT5A receptor. Here, we show that two different missense mutations in WNT5A, which result in amino acid substitutions of highly conserved cysteines, are associated with autosomal dominant Robinow syndrome. One mutation has been found in all living affected members of the original family described by Meinhard Robinow and another in a second unrelated patient. These missense mutations result in decreased WNT5A activity in functional assays of zebrafish and Xenopus development. This work suggests that a WNT5A/ROR2 signal transduction pathway is important in human craniofacial and skeletal development and that proper formation and growth of these structures is sensitive to variations in WNT5A function.
November 1973 · American Journal of Diseases of Children (1960)
© 2008-2021 ResearchGate GmbH. All rights reserved.
Uncut Cock
Free Porn Pussy
The Best Xxx
Sex Video Https
Milf 2021



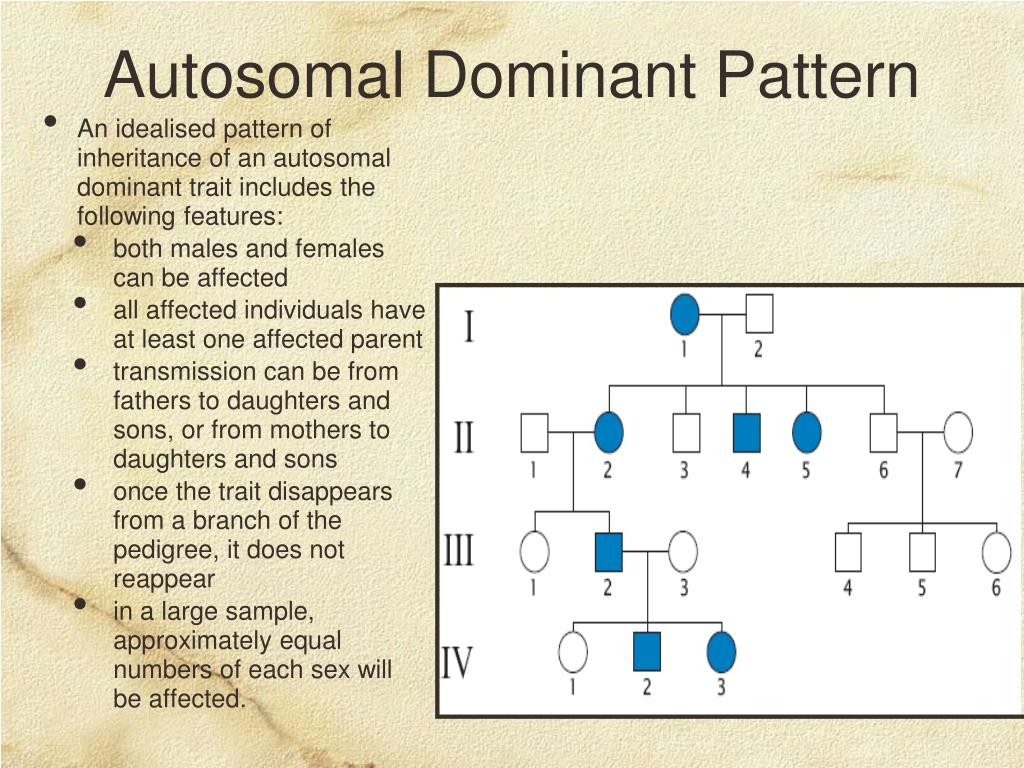
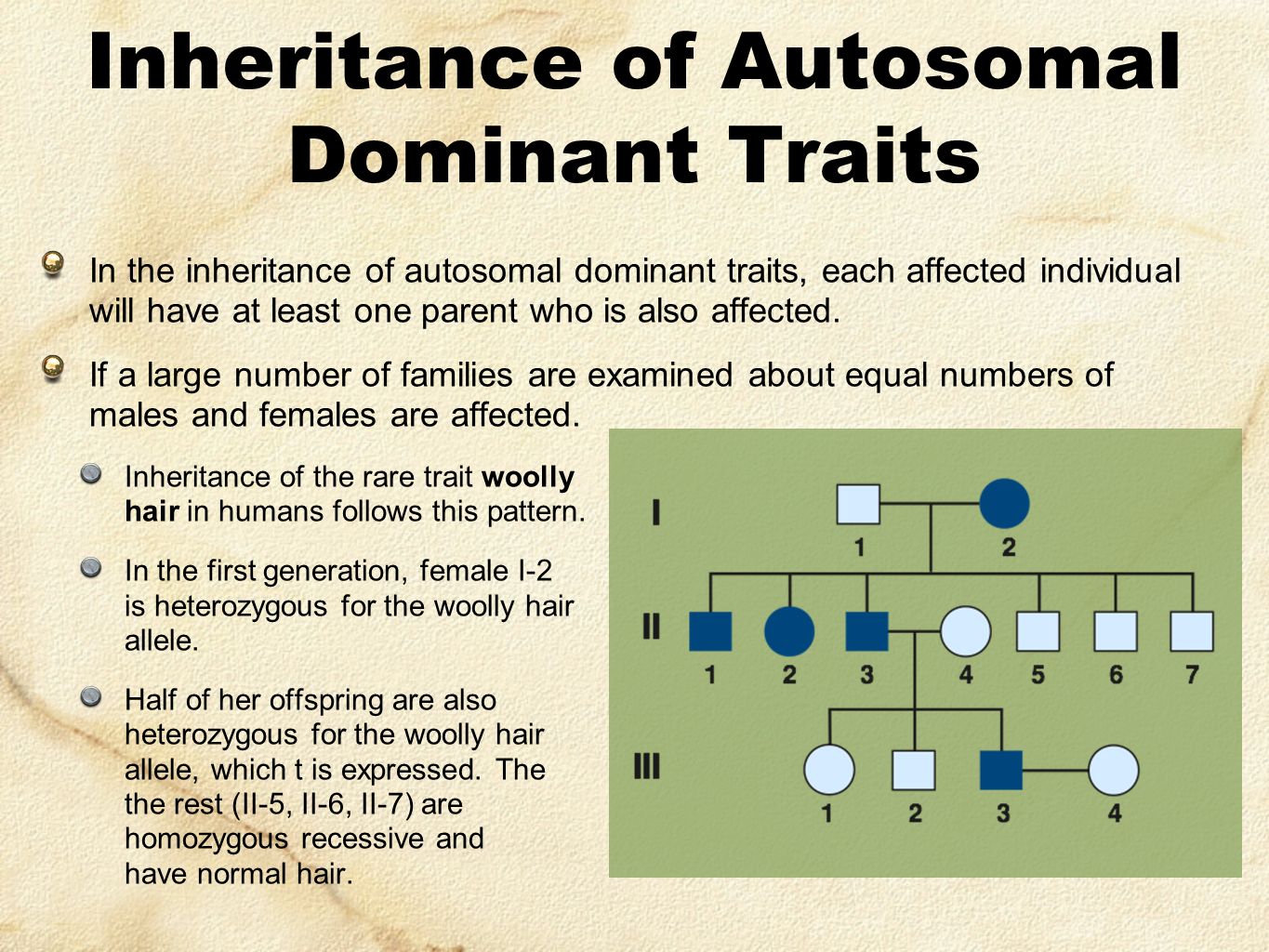

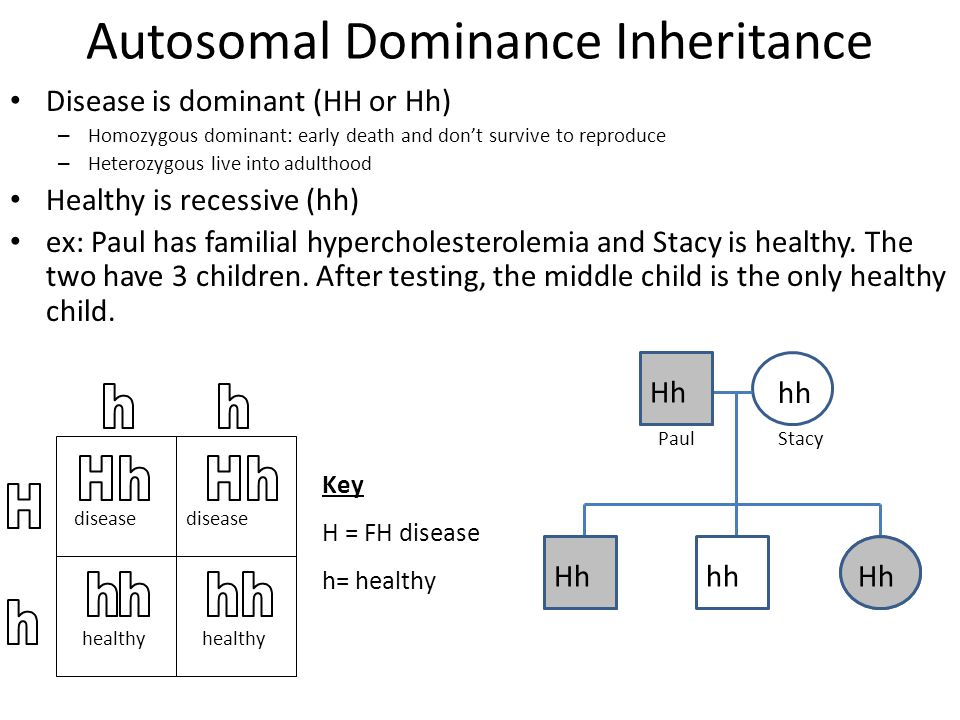
.jpg)
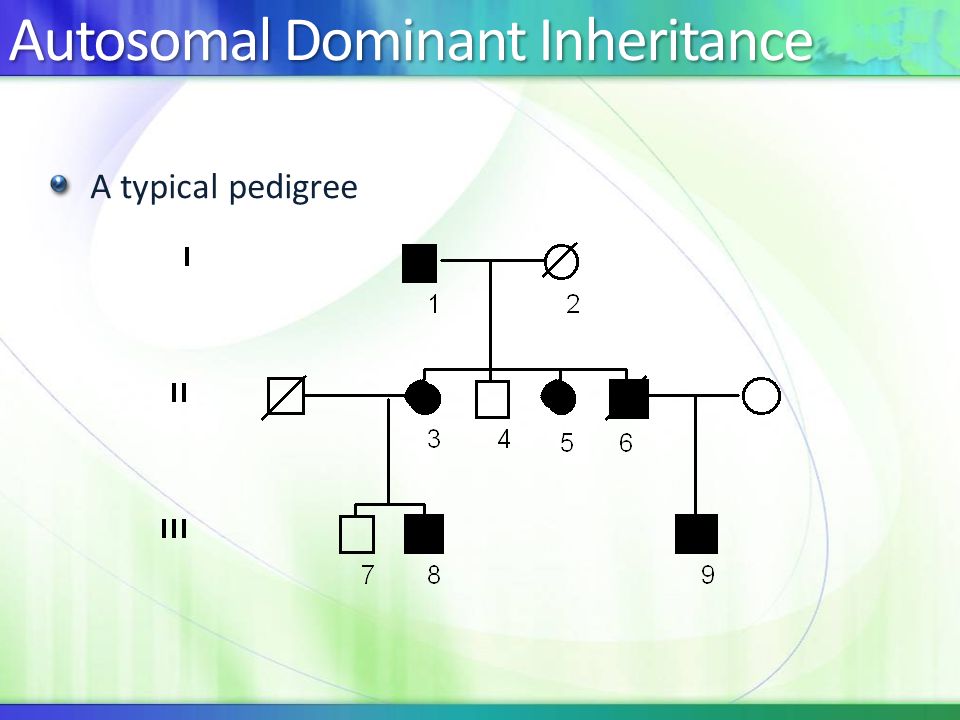
.jpg)