Homología de MSH3 y posible vínculo de recombinación con el sitio de escisión de furina del SARS-CoV-2
Balamurali K. AmbatiFrente. Virol., 21 de febrero de 2022 | https://doi.org/10.3389/fviro.2022.834808
Entre las numerosas diferencias de mutación puntual entre el SARS-CoV-2 y el coronavirus RaTG13 de murciélago, solo el sitio de escisión de furina (FCS) de 12 nucleótidos supera los 3 nucleótidos. Una búsqueda BLAST reveló que una porción de 19 nucleótidos del genoma SARS.Cov2 que abarca el sitio de escisión de la furina es una coincidencia 100% complementaria con una secuencia patentada optimizada por codón que es el complemento inverso del homólogo mutS humano (MSH3). La secuencia del complemento inverso presente en el SARS-CoV-2 puede ocurrir al azar, pero se deben considerar otras posibilidades. La recombinación en un huésped intermedio es una explicación poco probable. Los virus de ARN monocatenario, como el SARS-CoV-2, utilizan plantillas de ARN de cadena negativa en las células infectadas, lo que podría conducir a través de la recombinación de elección de copia con un ARN del SARS-CoV-2 de sentido negativo a la integración de la cadena negativa de MSH3, incluyendo el FCS, en el genoma viral. En cualquier caso, la presencia de la secuencia de ARN de 19 nucleótidos de largo que incluye el FCS con una identidad del 100% con el complemento inverso del ARNm de MSH3 es muy inusual y requiere más investigaciones.
Introducción
Con base en una publicación reciente que describe variantes de inserción de SARS-CoV-2 ( 1 ), nos gustaría llamar la atención sobre nuestros hallazgos recientes relacionados con la secuencia del sitio de escisión de furina (FCS) en SARS-CoV-2 Spike (S) proteína. El SARS-CoV-2 causante de la pandemia de COVID-19 ( 2) tiene una identidad de aminoácidos del 82,3 % con el coronavirus de murciélago SL-CoVZC45, una identidad de aminoácidos del 77,2 % con el SARS-CoV y una identidad de secuencia del genoma del 96,2 % con el coronavirus de murciélago RaTG13. Si bien existen numerosas diferencias de mutación puntual entre el SARS-CoV-2 y RaTG13, solo una inserción y una diferencia superior a 3 nucleótidos (nt): una inserción de 12 nucleótidos que codifica cuatro aminoácidos (aa 681-684, PRRA) en el SARS-CoV -Se ha descubierto la proteína 2 S. Este FCS polibásico diferencia al SARS-CoV-2 de otros betacoronavirus de linaje b o cualquier otro sarbecovirus ( 3 ). Una adición de FCS mejoró la infectividad del SARS Co-V-2 en 2019 ( 4 ). La ausencia de este FCS da como resultado variantes atenuadas del SARS-CoV-2 útiles para la vacunación animal, lo que acentúa su relevancia para la infección humana ( 5). Este FCS es vital para la transmisión entre humanos y hurones ( 6 ), expande el tropismo viral a las células humanas ( 7 ) y es un requisito para la enfermedad grave en dos modelos animales de SARS-CoV-2 ( 8 ).
Proteína de pico de SARS-CoV-2 y MSH3
Una característica peculiar de la secuencia de nucleótidos que codifica el sitio de escisión de furina PRRA en la proteína S del SARS-CoV-2 son sus dos codones CGG consecutivos. Este codón de arginina es raro en los coronavirus: el uso relativo de codones sinónimos (RSCU) de CGG en pangolín CoV es 0, en murciélago CoV 0,08, en SARS-CoV 0,19, en MERS-CoV 0,25 y en SARS-CoV-2 0,299 ( 9 ).
Una búsqueda BLAST de la inserción de 12 nucleótidos nos llevó a una coincidencia inversa del 100 % en una secuencia patentada (SEC ID 11652, nt 2751-2733) encontrada en la patente de EE. UU. 9 587 003 presentada el 4 de febrero de 2016 ( 10 ) ( Figura 1 ) . El examen de SEQ ID11652 reveló que la coincidencia se extiende más allá de la inserción de 12 nucleótidos a una secuencia de 19 nucleótidos:
5′-CTACGTGCCCGCCGAGGAG-3′ (nt 2733-2751 de SEQ ID11652), de modo que el ARNm resultante tendría 3′- GAUGCACGGGCGGCUCCUC -5′, o equivalentemente 5′- CU CCU CGG CGG GCA CGU AG-3′ (nucleótidos 23547-23565 en el genoma del SARS-CoV-2, en el que los cuatro codones en negrita producen PRRA, aminoácidos 681–684 de su pico proteína). Esto es muy raro en la base de datos NCBI BLAST.
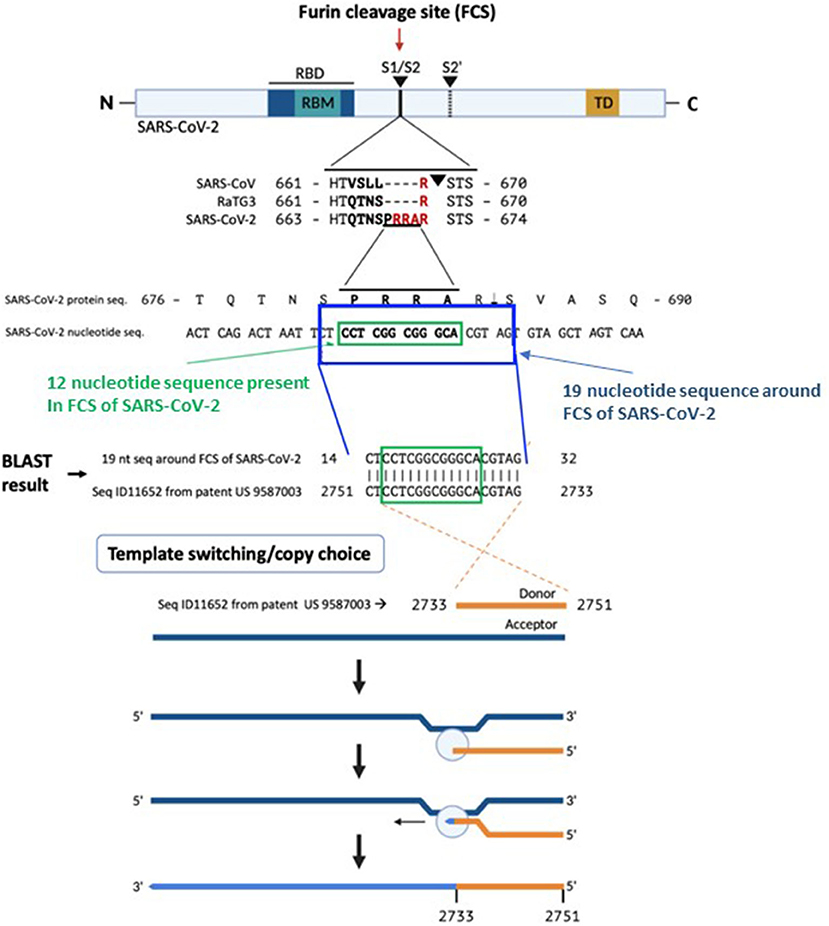
Figura 1 . El origen de la secuencia de furina en el SARS-CoV-2. Comparación de las secuencias de proteínas en la unión S1/S2 en SARS-CoV, RaTG13 y SARS-CoV-2 que demuestra la presencia del sitio de escisión de furina (FCS) PRRA solo en SARS-CoV-2. En base a una búsqueda BLAST del tramo de 12 nucleótidos que codifica FCS PRRA, se identificó una secuencia idéntica de 19 nucleótidos de largo en la secuencia patentada (US 958 7003) Seq ID11652. SEQ ID11652 se transcribe a un ARNm de MSH3 que parece tener codones optimizados para humanos. Esta secuencia de 19 nucleótidos, incluidos 12 nucleótidos que codifican FCS PRRA, presente en el gen MSH3 humano, podría haberse introducido en el genoma del SARS-CoV-2 mediante el mecanismo de recombinación de elección de copia ilustrado en células humanas infectadas con SARS-CoV-2 que sobreexpresan el gen MSH3.
La correlación entre esta secuencia de SARS-CoV-2 y el complemento inverso de una secuencia patentada de ARNm es de origen incierto. El análisis bioestadístico convencional indica que la probabilidad de que esta secuencia esté presente aleatoriamente en un genoma viral de 30.000 nucleótidos es de 3,21 × 10-11 ( Figura 2 ).

Figura 2 . Cálculos de la probabilidad de ocurrencia natural de la secuencia de 19nt en estudio. El genoma del SARS-CoV-2 tiene una longitud de ~30 000 nucleótidos (P1). La secuencia patentada tiene una longitud de ~3300 nucleótidos (P2). La biblioteca patentada abarca 24.712 secuencias de diferentes longitudes, con una longitud media en el rango de 3.300 nucleótidos. Se dan cálculos de probabilidad convencionales de la probabilidad de presencia de una secuencia de 19 nucleótidos en el genoma humano y en una de las secuencias de la biblioteca patentada.
La secuencia patentada SEQ ID11652, leída en la dirección de avance, codifica un 100 % de coincidencia de aminoácidos con el homólogo 3 de mut S humano (MSH3) ( 9 ). MSH3 es una proteína reparadora de desajustes de ADN (parte del complejo beta MutS) ( 11 ). SEQ ID11652 se transcribe a un ARNm de MSH3 que parece tener codones optimizados para humanos ( 12 ). No encontramos la secuencia de 19 nucleótidos CTCCTCGGCGGGCACGTAG en ningún genoma eucariótico o viral, excepto SARS-CoV-2 con una cobertura e identidad del 100 % en la base de datos BLAST ( Tablas complementarias 1 a 3 ).
Discusión
El reemplazo de MSH3 con una secuencia de ARNm optimizada por codón para la expresión humana probablemente tenga aplicaciones en cánceres con deficiencias en la reparación de errores de emparejamiento. Si bien una parte de una secuencia de complemento inverso que está presente en el SARS-CoV-2 podría ser una coincidencia aleatoria, otras posibilidades merecen consideración.
Se sabe que la sobreexpresión de MSH3 interfiere con la reparación del desajuste (el secuestro de MSH2 del complejo MutS alfa que comprende MSH2 y MSH6 da como resultado la degradación de MSH6 y el agotamiento de MutS alfa) ( 13 ), lo que tiene importancia virológica. La inducción de la deficiencia en la reparación del desajuste del ADN da como resultado la permisividad de la infección por el virus de la influenza A (IAV) de las células respiratorias humanas y una mayor patogenicidad ( 14 ). La deficiencia en la reparación de desajustes puede prolongar la eliminación del SARS-CoV-2 ( 15 , 16 ).
La ausencia de CTCCTCGGCGGGCACGTAG de cualquier genoma eucariótico o viral en la base de datos BLAST hace que la recombinación en un huésped intermedio sea una explicación poco probable de su presencia en el SARS-CoV-2.
Un ARNm optimizado con codón humano que codifica una proteína 100 % homóloga a la MSH3 humana podría, durante el curso de la investigación viral, inducir inadvertida o intencionalmente una deficiencia en la reparación de errores de emparejamiento en una línea celular humana, lo que aumentaría la susceptibilidad a una infección viral similar al SARS. La infección de células humanas transducidas con SEQ ID11652-MSH3 por un virus similar al SARS podría permitir la recombinación de elección de copia ( 15 ).
La replicación del SARS-CoV-2 y otros virus de ARN monocatenario con un genoma de ARN de polaridad positiva se inicia mediante la síntesis de ARN de cadena negativa en el citoplasma de las células infectadas ( 17 ) (Figura 1 ). El ARN de cadena negativa es una plantilla para la síntesis de ARN de cadena positiva utilizado para la traducción de proteínas no estructurales, el complejo de replicación y transcripción o nuevas cápsidas de virión. Los coronavirus generan ARN de doble cadena en una etapa temprana de la infección a través de la replicación genómica y la transcripción del ARNm ( 18 ).
La adquisición de la secuencia FCS del complemento inverso de un ARNm de MSH3 de sentido positivo sobreexpresado podría ocurrir a través de la recombinación de elección de copia con un ARN intermedio de SARS-CoV-2 de sentido negativo ( 15 ), lo que implica saltar de una plantilla a otra ( 19 ) ( Figura 1 ). La homología entre el SARS-CoV-2 y otros coronavirus conocidos se interrumpe y la mayoría de las secuencias del SARS-CoV-2 derivan de un ancestro común relativamente reciente con el murciélago RaTG13. Además, los gráficos de similitud (SimPlots) han identificado cambios repentinos en la identidad de secuencia entre SARS-CoV-2 y RaTG13, lo que indica posibles eventos de recombinación, lo que podría explicar la capacidad de unión de SARS-CoV-2 a ACE2 a través de su RBD, que no es el estuche para el RaTG13 RBD ( 15 ).
Una crítica a esta hipótesis es que la secuencia identificada está en la hebra opuesta del marco de lectura abierto en SEQ ID11652. Sin embargo, las células transfectadas con MSH3, que inducen la deficiencia en la reparación de errores de apareamiento, podrían haberse dirigido al ADNc de doble cadena que codifica la SEQ ID11652. Estas células cotransfectadas con un virus similar al SARS que expresa RdRp podrían unirse a esta secuencia de 19 nucleótidos ( 15 ) y permitir la integración de un fragmento de la cadena negativa en el genoma viral, incluido el FCS, a pesar de estar en la cadena opuesta de el marco de lectura abierto. Los mecanismos de reparación de desajustes han permitido la integración de fragmentos cortos de cadenas antisentido en modelos experimentales ( 20 , 21). La microhomología puede dirigir la recombinación entre MSH3 y un virus similar al SARS, que podría tener lugar en la secuencia de interés de 19 nucleótidos.
La presencia en el SARS-CoV-2 de una secuencia de ARN de 19 nucleótidos que codifica un FCS en el aminoácido 681 de su proteína espiga con un 100 % de identidad con el complemento inverso de una secuencia patentada de ARNm de MSH3 es muy inusual. Las posibles explicaciones de esta correlación deben investigarse más a fondo.
Material Suplementario
https://www.frontiersin.org/articles/10.3389/fviro.2022.834808/full#supplementary-material
Referencias
References
1. Garushyants SK, Rogozin IB, Koonin EV. Template switching and duplications in SARS-CoV-2 genomes give rise to insertion variants that merit monitoring. Commun Biol. (2021) 4:1343. doi: 10.1038/s42003-021-02858-9
PubMed Abstract | CrossRef Full Text | Google Scholar
2. Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, et al. A new coronavirus associated with human respiratory disease in China. Nature. (2020) 579:265–9. doi: 10.1038/s41586-020-2008-3
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Coutard B, Valle C, de Lamballerie X, Canard B, Seidah NG, Decroly E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res. (2020) 176:104742. doi: 10.1016/j.antiviral.2020.104742
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. (2020) 181:281–92.e286. doi: 10.1016/j.cell.2020.02.058
PubMed Abstract | CrossRef Full Text | Google Scholar
5. Lau SY, Wang P, Mok BWY, Zhang AJ, Chu H, Lee ACY, et al. Attenuated SARS-CoV-2 variants with deletions at the S1/S2 junction. Emerg Microbes Infect. (2020) 9:837–42. doi: 10.1080/22221751.2020.1756700
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Peacock TP, Goldhill DH, Zhou J, Baillon L, Frise R, Swann OC, et al. The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. Nat Microbiol. (2020) 6:899–909. doi: 10.1038/s41564-021-00908-w
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Xia S, Lan Q, Su S, Wang X, Xu W, Liu Z, et al. The role of furin cleavage site in SARS-CoV-2 spike protein-mediated membrane fusion in the presence or absence of trypsin. Signal Transd. Targeted Ther. (2020) 5:92. doi: 10.1038/s41392-020-0184-0
PubMed Abstract | CrossRef Full Text | Google Scholar
8. Hou W. Characterization of codon usage pattern in SARS-CoV-2. Virology J. (2020) 17:138. doi: 10.1186/s12985-020-01395-x
PubMed Abstract | CrossRef Full Text | Google Scholar
9. Kandeel M, Ibrahim A, Fayez M, Al-Nazawi M. From SARS and MERS CoVs to SARS-CoV-2: Moving toward more biased codon usage in viral structural and nonstructural genes. J Med Virol. (2020) 92:660–6. doi: 10.1002/jmv.25754
PubMed Abstract | CrossRef Full Text | Google Scholar
10. Bancel S, Chakraborty T, De Fougerolles A, Elbashir SM, John M, Roy A, et al. Modified Polynucleotides for the Production of Oncology-Related Proteins and Peptides. Cambridge, MA: United States Patent. (2016).
11. MacRae SL, McKnight Croken M, Calder RB, Aliper A, Milholland B, White RR, et al. DNA repair in species with extreme lifespan differences. Aging. (2015) 7:1171–84. doi: 10.18632/aging.100866
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Mauro VP, Chappell SA. A critical analysis of codon optimization in human therapeutics. Trends Mol Med. (2014) 20:604–13. doi: 10.1016/j.molmed.2014.09.003
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Marra G, Iaccarino I, Lettieri T, Roscilli G, Delmastro P, Jiricny J. Mismatch repair deficiency associated with overexpression of the MSH3 gene. Proc Natl Acad Sci USA. (1998) 95:8568. doi: 10.1073/pnas.95.15.8568
PubMed Abstract | CrossRef Full Text | Google Scholar
14. Chambers BS, Heaton BE, Rausch K, Dumm RE, Hamilton JR, Cherry S, et al. DNA mismatch repair is required for the host innate response and controls cellular fate after influenza virus infection. Nat Microbiol. (2019) 4:1964–77. doi: 10.1038/s41564-019-0509-3
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Gallaher WR. A palindromic RNA sequence as a common breakpoint contributor to copy-choice recombination in SARS-COV-2. Arch Virol. (2020) 165:2341–8. doi: 10.1007/s00705-020-04750-z
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Haque F, Lillie P, Haque F, Maraveyas A. Deficient DNA mismatch repair and persistence of SARS-CoV-2 RNA shedding: a case report of Hereditary Nonpolyposis Colorectal Cancer with COVID-19 infection. BMC Infect Dis. (2020) 21:854. doi: 10.1186/s12879-021-06500-1
PubMed Abstract | CrossRef Full Text | Google Scholar
17. V'kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev. (2021) 19:155–70. doi: 10.1038/s41579-020-00468-6
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Graham RL, Baric RS. Recombination, reservoirs, and the modular spike: mechanisms of coronavirus cross-species transmission. J Virol. (2010) 84:3146. doi: 10.1128/JVI.01394-09
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Sola I, Almazan F, Zuniga S, Enjuanes L. Continuous and discontinuous RNA synthesis in coronaviruses. Annu Rev Virol. (2015) 2:265–88. doi: 10.1146/annurev-virology-100114-055218
PubMed Abstract | CrossRef Full Text | Google Scholar
20. Rakosy-Tican E, Lörincz-Besenyei E, Molnár I, Thieme R, Hartung F, Sprink T, et al. New Phenotypes of potato co-induced by mismatch repair deficiency and somatic hybridization. Front Plant Sci. (2019) 10:3. doi: 10.3389/fpls.2019.00003
PubMed Abstract | CrossRef Full Text | Google Scholar
21. Harmsen T, Klaasen S, van de Vrugt H, te Riele H. DNA mismatch repair and oligonucleotide end-protection promote base-pair substitution distal from a CRISPR/Cas9- induced DNA break. Nucl Acids Res. (2018) 46:2945–55. doi: 10.1093/nar/gky076
PubMed Abstract | CrossRef Full Text | Google Scholar