DICER1-синдром
HSO talks
Автор: Влада Богородова
Редакция: Даша Моргачева
В детской онкологии есть много наследственных синдромов предрасположенности к опухолевым заболеваниям, о которых не всегда знают даже специалисты нашего профиля. Так что сегодня начнем повышать свой уровень осведомленности (и ЧСВ, конечно) и поговорим об одном из таких заболеваний — DICER1 синдроме.
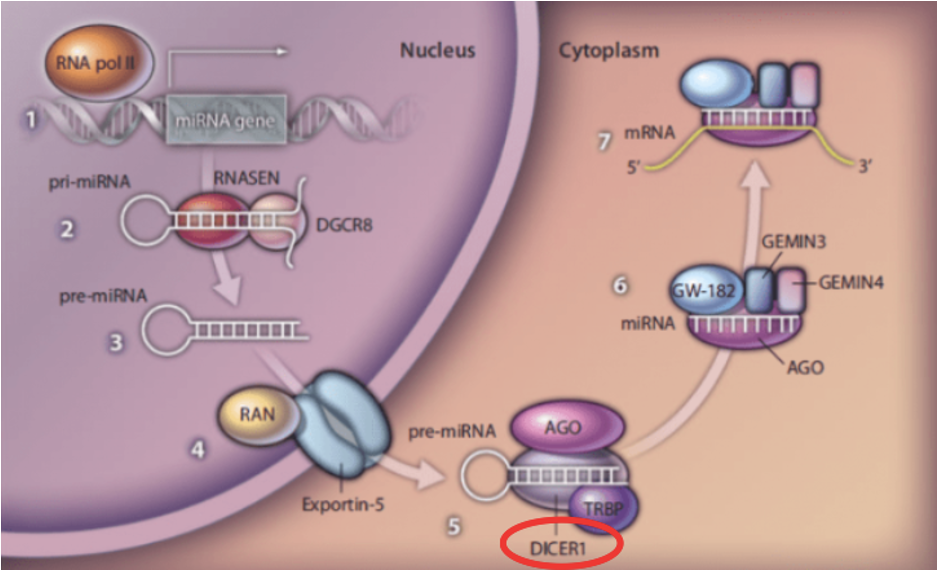
DICER1 синдром — это заболевание с аутосомно-доминатным типом наследования, которое возникает вследствие мутации в одноименном гене. Ген кодирует эндорибонуклеазу Dicer, ответственную за выработку зрелых микроРНК, которые принимают участие в транскрипционной и посттранскрипционной регуляции экспрессии генов.
При DICER1 синдроме происходит мутация в одноименном гене, что, нарушает процессинг микроРНК и вызывает изменение регуляции экспрессии генов. Болеют в основном дети, у взрослых синдром встречается крайне редко.
К заболеваниям, возникающим при DICER1 синдроме, относятся:
- плевропульмональная бластома (ППБ)
- кистозная нефрома (КН)
- многоузловой зоб (МУЗ)
- опухоли яичников из клеток Сертоли–Лейдига
- лимфома Ходжкина, пинеобластома и другие
Давайте чуть подробнее поговорим о наиболее характерных для DICER1 синдрома опухолях.
Плевропульмональная бластома
ППБ — редкая мезенхимальная опухоль легких и/или плевры, одна из самых распространенных при DICER1 синдроме. Впервые заболевание было описано в 1952 году Barnard как «эмбриома» легкого, основываясь на гистологическом подобии опухоли и эмбриональной ткани легкого плода.
Термин «бластома» был введен позже, в 1961 году Spencer, который предположил происхождение опухоли из мезодермальной бластемы, аналогично таким детским опухолям, как нефробластома, нейробластома, гепатобластома и др.
Выделяют 3 типа ППБ:
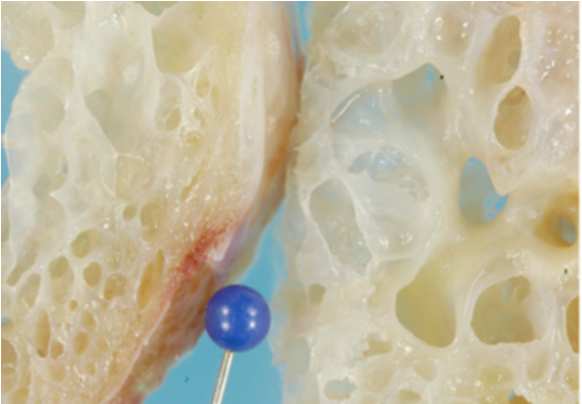
- Тип I характеризуется кистозными опухолями в легких, которые могут подвергаться малигнизации, но в целом имеют более благоприятный прогноз (5-летняя ОВ около 89%, если не происходит трансформации в типы II и III). Чаще всего возникает в возрасте < 2 лет, медиана — 10 месяцев. Выделяют еще подтип Ir, который также характеризуется кистозными образованиями, но может спонтанно регрессировать и не имеет злокачественный потенциал.
- Тип II — смешанные кистозные и солидные образования, медиана возраста 35 месяцев. 5-летняя ОВ достигает 74%.
- Тип III — исключительно солидные опухоли, медиана возраста 41 месяц. Этот тип наиболее агрессивный и даже при проведении комплексной терапии 5-летняя ОВ составляет около 50%.

Типы II и III имеют метастатический потенциал, при этом чаще всего поражается головной мозг (в 11% случаев для типа II и 55% для типа III).
Клиническая картина
Клиническая картина ППБ, как и у большинства детских опухолей, неспецифична. Чаще всего бывают жалобы на боли в груди, кашель, повышение температуры, одышку, слабость (более характерно для II и III типов опухоли). Иногда заболевание манифестирует остро с клиникой дыхательной недостаточности вследствие напряженного пневмоторакса.
Лабораторные данные также неспецифичны, чаще всего в гемограмме отмечается тенденция к анемии, умеренный лейкоцитоз, тромбоцитоз.
Диагностика
Рентгенография легких не всегда позволяет выявить образования, поэтому золотым стандартом все же является КТ легких с контрастным усилением. В зависимости от типа ППБ картина может быть представлена множественными мультикистозными структурами, кистами с солидным компонентом или исключительно солидными мягкотканными образованиями.
Для окончательной верификации диагноза выполняется эндоскопическая торакоскопия с биопсией образований и последующим гистологическим и иммуногистохимическим исследованиями.
Лечение
Лечение подразумевает максимально возможную безопасную резекцию образований, при необходимости объем операции может быть расширен до лобэктомии или пневмонэктомии.
Типы II и III требуют проведения адъювантной химиотерапии, но учитывая редкую встречаемость опухоли стандарта лечения нет. Существуют разные подходы и режим введения препаратов, но базово используют винкристин, дактиномицин, ифосфамид/циклофосфамид, доксорубицин.
Эффективность лучевой терапии в лечении ППБ в клинических исследованиях не установлена, но ввиду агрессивности опухоли и высокого риска рецидива, может быть использована.
Существует международный реестр ППБ/DICER1 для пациентов и членов их семей. Целью реестра является регистрация клинических случаев и получение большего количества данных о течении, диагностике и терапии ППБ и DICER1-ассоциированных заболеваний: https://www.ppbregistry.org/
Кистозная нефрома
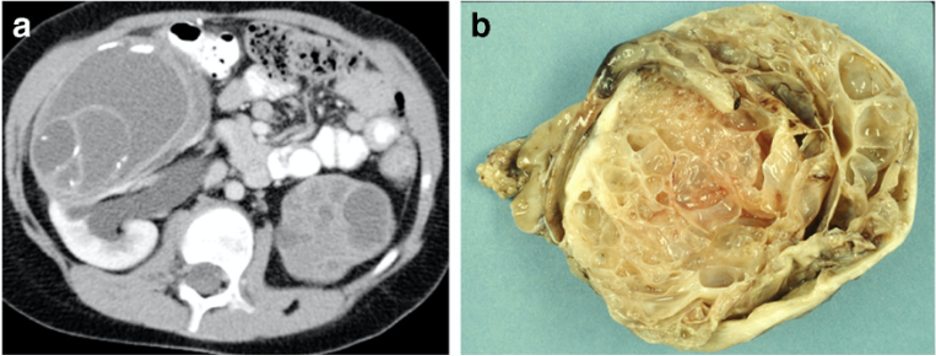
КН представляет собой доброкачественную мультикистозную опухоль почек и имеет 2 пика: около 65% случаев возникает в возрасте до 4 лет, еще 35% - между 40 и 60 годами.
Частота герминальной мутации гена DICER1 у пациентов с кистозной нефромой составляет до 73%. В редких случаях наблюдается трансформация кистозной нефромы в анапластическую саркому почки.
Клинически заболевание может проявляться гематурией, болью в боку и увеличением объема живота. Лечение подразумевает радикальную нефрэктомию или резекцию пораженной почки с удалением опухоли.
Описан случай, как у одного пациента, получавшего химиотерапию по поводу ППБ, был остановлен прогрессирующий рост двусторонних кист почек, что предотвратило необходимость последующей трансплантации.
Хотя это всего лишь единичный случай, он может свидетельствовать о положительной роли химиотерапии; однако в настоящее время недостаточно данных, чтобы обосновать какие-либо терапевтические рекомендации.
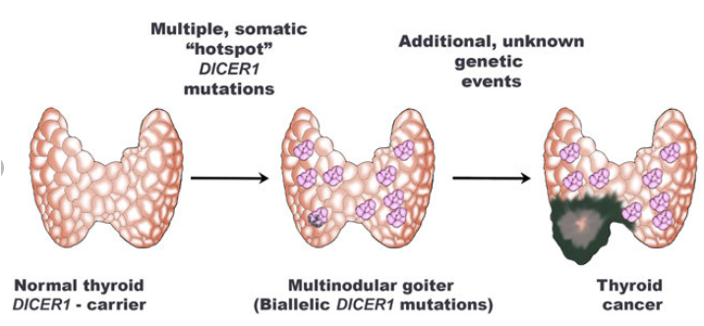
Многоузловой зоб
МУЗ проявляется возникновением множественных узловых образований щитовидной железы. В исследованиях было продемонстрировано, что у 75% женщин и 17% мужчин с синдромом DICER1 наблюдаются образования щитовидной железы, по сравнению с контрольной популяцией: 8% и 0% для женщин и мужчин соответственно.
Существует распространенное утверждение: большинство узлов щитовидной железы у детей, как и у взрослых пациентов, являются доброкачественными. Однако узлы, выявляемые у пациентов до 18 лет, в 3, а по некоторым данным и в 5 раз чаще оказываются злокачественными.
То есть у детей 20–30% узлов являются злокачественными по сравнению с 5–10% у взрослых.
У большинства пациентов-детей на момент обнаружения узлового зоба симптомы отсутствуют, и обнаруживается он либо во время медицинского осмотра, либо во время не связанного с обследованием щитовидной железы лучевого исследования головы и шейного отдела.
МУЗ и заболевания щитовидной железы распространены среди населения в целом, и поэтому синдром DICER1 следует рассматривать для пациентов с МУЗ только при наличии дополнительных опухолей, связанных с DICER1, у самого пациента или его родственников.
Рак щитовидной железы является редким проявлением синдрома DICER1 и может возникать вторично у детей, которые уже получали химио- и/или лучевую терапию по поводу предыдшествующего лечения.
При выявлении рака щитовидной железы выполняется тиреоидэктомия с последующей радиойодтерапией.
Опухоли из клеток Сертоли-Лейдига
Опухоли из клеток Сертоли-Лейдига - редкие новообразования яичников, которые могут возникать как у маленьких детей, так и у взрослых пациенток (от 2 до 40 лет).
Для этого типа опухолей характерна гиперандрогения, что приводит к вирилизации пациенток: появляются жалобы на аменорею, рост волос на лице и теле, угревые высыпания, гипертрофию клитора.
Хоть опухоли из клеток Сертоли-Лейдига редко встречаются среди всех новообразований яичников (<0,5%), недавнее исследование показало, что у 57% пациентов с данными опухолями были обнаружены герминальные мутации гена DICER1.
У пациенток с клиническими проявлениями вирилизации, обусловленными эндокринной секрецией опухоли, необходимо проведение исследования сывороточного уровня таких гормонов, как дегидроэпиандростерон, дегидроэпиандростерон- сульфат, 17-гидроксипрогестерон, кортизол. Это позволяет не только исключить надпочечниковый генез гиперандрогении, но и осуществить контроль эффективности лечения.
Единых стандартов ведения пациенток с опухолями из клеток Сертоли–Лейдига не существует. Основным методом лечения является хирургическое вмешательство, при этом объем его основывается на нескольких факторах, включающих стадию, степень дифференцировки опухоли и возраст пациентки.
В педиатрической практике, конечно, максимально предпочтительно провести органосохраняющую операцию, оставить второй яичник и матку, что позволит в будущем реализовать репродуктивную функцию пациентке.

Как обнаружить DICER1 синдром
Несмотря на повышенный риск возникновения злокачественных опухолей у пациентов с патогенными герминальными мутациями DICER1 гена, исследования демонстрируют, что к 50 годам опухоли возникают лишь у 19,3% пациентов с патогенными мутациями. На сегодняшний день существуют критерии для генетического консультирования DICER1 синдрома, которое может быть предложено пациентам с 1 большим или 2 малыми критериями:
- большие критерии: ППБ, легочные кисты в детском возрасте, грудная эмбриональная рабдомиосаркома, КН, мочеполовая саркома, овариальные опухоли из клеток Сертоли-Лейдига, гинандробластома, цервикальная или овариальная эмбриональная рабдомиосаркома, мочеполовые/гинекологические нейроэндокринные опухоли, МУЗ или рак щитовидной железы у 2-х и более родственников 1-й линии или у пациента с семейным анамнезом, предполагающим DICER1, МУЗ или дифференцированный рак щитовидной железы в детском возрасте, медуллоэпителиома, назальная хондромезенхимальная гамартома, пинеобластома, гипофизарная бластома.
- малые критерии: легочные кисты во взрослом возрасте, почечные кисты, опухоль Вилмса, МУЗ или дифференцированный рак щитовидной железы, эмбриональная рабдомиосаркома (кроме грудной и гинекологической локализации), низкодифференцированные нейроэндокринные опухоли, недифференцированная саркома, макроцефалия.
Для пациентов с уже установленным диагнозом DICER1 синдрома рекомендован скрининг в форме рентгенографии или КТ органов грудной клетки каждые 4-6 месяцев до 8 лет и каждые 12 месяцев до 12 лет.
Ссылки
- Caroleo AM, De Ioris MA, Boccuto L, et al. DICER1 Syndrome and Cancer Predisposition: From a Rare Pediatric Tumor to Lifetime Risk. Front Oncol. 2021;10:614541. Published 2021 Jan 21. doi:10.3389/fonc.2020.614541
- Schneider DT, Brecht IB, Olson TA, Ferrari A: Rare Tumors In Children and Adolescents, 2012
- Sabapathy, Divya G., et al. "Radiographic screening of infants and young children with genetic predisposition for rare malignancies: DICER1 mutations and pleuropulmonary blastoma." American Journal of Roentgenology 204.4 (2015): W475-W482.
- Bahubeshi A, Tischkowitz M, Foulkes WD. miRNA processing and human cancer: DICER1 cuts the mustard. Sci Transl Med. 2011 Nov 30;3(111):111ps46. doi: 10.1126/scitranslmed.3002493.
-Robertson J. C., Jorcyk C. L., Oxford J. T. DICER1 syndrome: DICER1 mutations in rare cancers //Cancers. – 2018. – Т. 10. – №. 5. – С. 143.