在SARS-CoV-2测序数据库中鉴定莫努匹拉韦相关的突变特征
https://doi.org/10.1101/2023.01.26.23284998
本文为预印本,未经同行评审。它报告了尚未评估的新医学研究,因此不应用于指导临床实践。
摘要
莫努匹拉韦是一种已广泛用于对抗SARS-CoV-2的抗病毒药物,通过在复制过程中诱导病毒基因组突变而起作用。大多数随机突变可能对病毒有害,许多突变将是致命的。莫努匹拉韦诱导的突变率升高已被证明可以降低动物模型中的病毒载量。然而,一些接受莫努匹拉韦治疗的患者可能无法完全清除SARS-CoV-2感染,并有可能继续传播莫努匹拉韦突变病毒。我们着手系统地研究全球测序数据库,以寻找莫努匹拉韦诱变的特征。我们发现,从2022年开始,在引入莫努匹拉韦治疗之后,以及在广泛使用该药物的国家和年龄组中,几乎只以序列出现一类特定的长系统发育分支。我们从莫努匹拉韦的AGILE安慰剂对照临床试验中计算了突变谱,并表明其特征,G-to-A和C-to-T速率升高,在很大程度上对应于这些长分支中看到的突变谱。我们的数据表明,在全球测序数据库中可以看到莫努匹拉韦诱变的特征,在某些情况下是向前传播的。
介绍
莫努匹拉韦是一种抗病毒药物,在一些国家获得许可用于治疗COVID-19。在体内,莫努匹拉韦最终转化为核苷酸类似物,三磷酸莫努匹拉韦(MTP)1.MTP能够在链合成过程中被掺入RNA中,特别是通过病毒RNA依赖性RNA聚合酶,在病毒基因组复制过程中可能导致序列保真度错误。RNA 复制中的这些错误导致许多病毒后代无法存活,因此降低了病毒的有效生长速度——莫努匹拉韦被证明可以在体外将 24 小时内的病毒复制减少 880 倍,并降低动物模型中的病毒载量(Rosenke 等人,2021 年)。莫努匹拉韦最初显示出治疗 COVID-19 的有限疗效(Jayk Bernal 等人,2022 年;Extance,2022 年),但随后更大规模的临床试验发现,莫努匹拉韦并没有降低高危人群的住院率或死亡率(Butler,2022 年)。作为市场上首批口服生物可利用的抗病毒药物之一,莫努匹拉韦已被许多国家广泛采用,最近一次是中国(路透社,2022 年)。然而,最近的试验结果和更有效的抗病毒药物的批准导致一些国家基于有限的有效性建议不要使用莫努匹拉韦(NICE指南;NC19CET,2022)。
MTP似乎主要通过作为胞嘧啶(C)的类似物,配对相反的鸟嘌呤(G)碱基(图1)而掺入新生RNA。然而,一旦掺入,莫努匹拉韦(M)碱可以转变为类似于尿嘧啶(U)的替代互变异构形式。这意味着在下一轮复制中,为了给出正义SARS-CoV-2基因组,M碱基可以与腺嘌呤(A)配对,导致G到A突变,如图2所示。MTP的掺入也可能发生在正义基因组的第二步合成过程中。在这种情况下,初始正义 C 在第一轮复制中与 G 正确配对,但在正义合成期间,该 G 与 M 碱基配对。在下一轮复制中,这个M可以与A配对,这将在最终的正义基因组中产生U,整个过程产生C到U突变(图)。S1)。
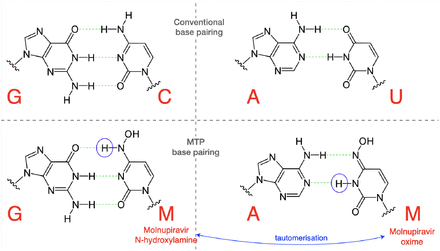
图1.三磷酸莫努匹拉韦可以采用多种互变异构形式。
N-羟胺形式类似于胞嘧啶,而肟形式更类似于尿嘧啶。因此,它们分别与鸟嘌呤和腺嘌呤配对。(图部分改编自马龙和坎贝尔 (2021) 。
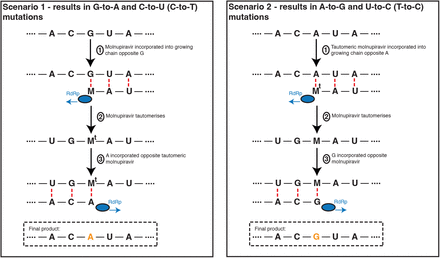
图2.莫努匹拉韦驱动G-to-A和C-to-U(C-to-T)突变,并在较小程度上驱动A-to-G和T-to-U(T-to-C)突变
在最常见的情况下,如左侧所示,M与G核苷酸相对。然后它可以在随后的复制中与A配对,产生G到A突变。如果原始G是由编码链C的负链合成引起的,那么G到A的变化最终会产生C到U编码的变化(图)。S1)。在右侧显示的第二种不太常见的场景中,M最初是通过与A配对而合并的,这可能导致A到G突变,或者,如果原始A来自编码U,则会导致U到C突变。
与掺入 RNA 时相比,游离核苷酸 MTP 更不容易发生互变异构化,因此突变的这种方向性是最有可能的(Gordon 等人,2021 年)。然而,一些MTP也可能代替U与A碱基结合,并反向经历上述过程,导致A到G和U到C突变(图2中的场景2,也是图)。S1)。
有人提出,许多主要的SARS-CoV-2变种是从长期慢性感染中出现的。该模型解释了变异的几个特点,例如普遍缺乏遗传中间体、生根于更老的序列、较长的系统发育分支长度以及与已知慢性感染的趋同进化水平(Rambaut 等人,2020 年;维亚纳等人,2022 年;希尔等人,2022 年;赫拉利等人,2022 年)。
在分析SARS-CoV-2树中的长系统发育分支时,已经注意到最近显示出莫努匹拉韦驱动诱变潜在特征的分支,包括指示向前传播的序列簇。因此,我们旨在系统地鉴定可能受莫努匹拉韦影响的序列,并表征其突变谱,以检查这些特征在全球测序数据库中的出现程度。
结果
为了更系统地研究这种模式,我们分析了来自McBroome等人(2021)的突变注释树,其中包含来自GISAID(易北河和巴克兰梅雷特,13)和INSDC数据库(Cochrane等人,2)的>2017万个SARS-CoV-2011序列。对于树的每个分支,我们计算了每个替换类的数量(A-to-T,A-to-G等 - 我们在本文的其余部分使用T而不是U,就像在测序数据库中一样)。将此树过滤为涉及至少 20 个替换的分支,并绘制替换类型的比例,揭示了该空间中具有较高 G 到 A 且几乎完全过渡替换的区域2,仅包含 2022 年采样的分支(图 3A),表明一些变化(无论是生物学还是技术)导致了新的突变特征。
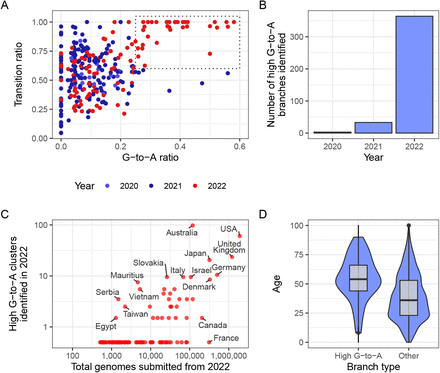
图3.2022年,全球测序数据库中一些国家出现了具有高G-A和高转换比的新突变特征
(A) 此散点图中的每个点代表突变注释树的一个分支,具有 >20 个替换。点根据分支突变的比例定位,这些突变是G到A(x)或任何过渡突变(y),并按它们发生的年份着色。具有较高 G 到 A 且几乎完全过渡突变的盒装区域仅在 2022 年出现。(B)满足特定标准的分支数量计数(G--A比>= 25%,C-T比>= 20%过渡比>95%,总突变>10)。(C)总基因组数量与已确定的高G-to-A簇数量的比较(使用与B相同的标准)。注意对数轴(半对数,截断线表示零)。例如,澳大利亚有来自总共97,119个基因组的194个簇,而法国有来自0,313个基因组的680个簇。(D)比较来自美国的>10个突变的集群的年龄分布,根据它们是否满足高G-to-A标准进行划分。高 G 到 A 集群对应于老年人。
注意到这种特征也涉及高比例的C-to-T突变,我们为感兴趣的分支创建了一个标准,我们称之为“高G-toA”分支:我们选择了至少涉及10个替换的分支,其中至少25%是G到A,至少20%是C到T,最多5%是转位。同样,这些分支在 2022 年几乎全部采样(图 3B)。这些分支主要从少数国家取样,这不能用测序工作的差异来解释(图3C,表1)。许多G-to-A分支比例较高的国家使用莫努匹拉韦:截至380年底,澳大利亚有>000,2022个处方(老年护理部网络研讨会,2022年),英国有>30,000个(NHS,2023年;巴特勒,2022 年),以及 240 年前几个月在美国的 >000,2022 人(Gold 等人,2022 年)。全测序水平高但G-to-A分支数量较少的国家(加拿大、法国)尚未授权使用莫努匹拉韦(加拿大政府,2022 年;斯宾塞等人,2021 年)。来自美国的年龄元数据显示,与具有相似数量突变但没有选择替代类型的对照分支相比,这些高G-to-A分支对年龄较大的患者存在显着偏倚(图.3D)。在澳大利亚有年龄数据的地方,它还确定了主要在老年人口中的长分支。这与这些国家优先使用莫努匹拉韦治疗老年人是一致的,老年人的严重感染风险更大。在澳大利亚,莫努匹拉韦被预先放置在老年护理机构中,建议所有 70 岁或以上的患者,无论有无症状,都应考虑使用莫努匹拉韦(澳大利亚卫生和老年护理部,2022 年)。
内联查看
表 1.25 年按国家/地区确定的高 G 到 A 数量(G 与 A 比率 >= 20%,C 与 T 比率 >= 95% 过渡比> 10%,总突变> 2022)与基因组总数。仅包括拥有>10,000个基因组的国家。
我们发现,高G-to-A分支的分支长度分布与其他类型的分支不同,分支长度较长时富集(图4)。接下来,我们试图看看这些高G-to-A分支是否与类似长度的其他分支不同的突变率相关。我们使用 Chronumental (Sanderson,2021 年)为树的每个节点分配了一个日期,并观察到具有高 G-toA 签名的分支在时间上测量的分支长度比没有此签名的匹配长度的分支短(图)。S2),提示突变率增加。

图4.与其他分支相比,高 G 到 A 分支的分支长度分布
为了对分支类型充满信心,该图仅限于大于 10 的分支长度,并且由于从 UShER MAT 中排除了长分支样本,因此分支长度限制为小于 20。 分支被过滤到 2022 年的分支。
在测序数据库中,采样的大多数长分支只有一个后代尖端序列,但在某些情况下,分支已经产生了具有大量后代序列的集群。例如,2022 年 20 月澳大利亚的一个集群涉及 5 个尖端序列,具有不同的年龄元数据表明它们确实来自多个个体(图 25)。该集群涉及主分支中的44个替换,其中所有替换都是转换,36%是C到T,2022%是G到A。密切相关的外群可追溯到 1 年 2 月,这表明这些突变在 6-<> 个月内出现。还有许多其他具有多个后代序列的高G到A分支的例子,包括来自英国(图<>A,B)。
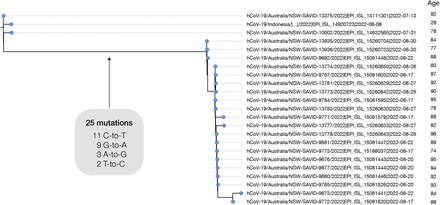
图5.从高G到A突变事件中出现的20个个体的集群
该集群涉及在可能不到一个月内发生的25个突变的盐化,所有这些都是过渡替代,G-to-A率升高。序列用年龄元数据注释,提示老年护理机构中暴发。
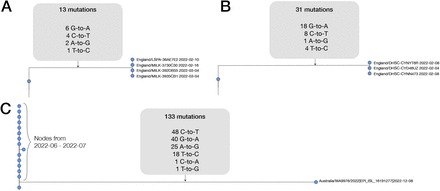
图6.三个高G-to-A事件的系统发育树
(A) 2022 年 13 月至 2022 月来自英国的 31 个序列集群,其中 133 个具有高 G 到 A 特征的共享基因。(B) 从 2 年 133 月起来自英国的四个序列集群,其中 <> 个具有高 G-to-A 特征的共享基因。(C)来自澳大利亚的单例序列,具有高G-to-A特征和总共<>个突变。观察到的<>个突变中只有<>个是转位,并且过渡包括许多G到A事件。
在构建每日更新的突变注释树(McBroome 等人,2021 年)的过程中,排除了与现有树高度不同的样本。鉴于某些SARS-CoV-2测序数据中的技术错误,这是一个必要的步骤,但这也意味着高度不同的莫努匹拉韦诱导的序列可能会被排除在外。为了研究这种效应,我们为澳大利亚创建了一个全面的突变注释树,甚至可以识别该子集中最不同的序列。该分析允许识别涉及多达130个替换的突变事件(图6C,图)。S3),具有相同的特征,即G-to-A突变率升高,几乎完全是过渡替换。我们确定的具有这些非常高数量的突变的病例涉及单个序列,并且可能代表由接受多个疗程莫努匹拉韦治疗的慢性感染个体产生的序列。
接下来,我们评估了这些分支上的突变谱(上下文核苷酸替换的模式)。我们确定的谱系(图7A)以G到A和C到T过渡突变为主,A到G和T到C转换的贡献较小。这种模式与莫努匹拉韦的已知作用机制一致(图2)。这些转变表现出对特定周围核苷酸环境的偏好,例如G-to-A突变最常见于TGT和TGC环境。这可能代表病毒RdRp倾向于与特定周围核苷酸结合,病毒RdRp倾向于将莫努匹拉韦与特定核苷酸相邻,或者倾向于在特定上下文环境中使用病毒校对核酸外切酶去除莫努匹拉韦。
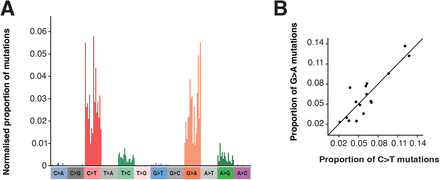
图7.全球测序数据库中长系统发育分支的突变谱
(A)长系统发育分支的单碱基取代(SBS)谱。(B)长系统发育分支上C-to-T和G-to-A突变的上下文突变偏好比较。绘制了等效(反向)环境中 C 到 T 或 G 到 A 突变的比例。存在显着的相关性,显示出对C-to-T和G-to-A突变的相似上下文偏好。比例根据上下文在基因组中出现的次数进行归一化。
C-to-T和G-to-A突变在光谱中的主导地位可能是由于莫努匹拉韦诱导的G-toA突变在病毒复制过程中合成不同链。在负链合成过程中掺入莫努匹拉韦将导致病毒共识序列中的G-to-A突变,而在正链合成过程中的掺入将被视为C-to-T突变(图)。S1)。与此一致,我们观察到C-to-T和G-to-A突变中等效上下文模式的突变偏差之间存在很强的正相关关系(Pearson's r = 0.88,95%CI 0.68-0.96,p < 0.001,图7B)(例如,一条链上ACG环境中的C-to-T突变等同于另一条链上CGT上下文中的G-to-A突变)。
为了将观察到的特征与已知莫努匹拉韦暴露个体的突变进行比较,我们重新检查了来自 AGILE IIa 期临床试验的基因组数据集(Donovan-Banfield 等人 (2022),NCT04746183)。我们首次通过比较同一患者的第1天样本(在治疗开始前采集)和第5天样本来分析可能的莫努匹拉韦诱导的突变谱。该数据集的优点是它还包括接受安慰剂治疗的个体,在没有莫努匹拉韦的情况下提供突变光谱对照。我们发现,莫努匹拉韦治疗的患者表现出明显高于安慰剂治疗的患者的突变负荷(方差分析p < 0.001,图)。S6)。安慰剂和莫努匹拉韦之间的突变谱差异很大(图8A,光谱之间的余弦相似性= 0.68)。假设安慰剂患者内的突变过程也发生在莫努匹拉韦治疗中,我们减去安慰剂谱以获得莫努匹拉韦特异性诱导的突变(图8A)。同样,我们发现转换突变的显着富集(图8B-C)。

图8.来自AGILE临床试验的莫努匹拉韦突变谱
(A)在AGILE试验中接受安慰剂或莫努匹拉韦治疗的患者的SBS突变谱。右图显示了莫努匹拉韦诱导的突变,通过从莫努匹拉韦治疗患者的所有突变谱中减去安慰剂谱(左图)来计算(中图)。光谱显示为每位患者每个可用上下文的突变数。(B)安慰剂和莫努匹拉韦治疗之间每种突变类型的数量(在所有情况下求和)的比较。误差线显示突变自举的置信区间(参见方法)。(C)通过将莫努匹拉韦的突变负荷除以安慰剂的突变负荷来计算治疗之间每种突变类型的比率。红色虚线表示相同的负担。
每个过渡突变中的上下文模式在已知的莫努匹拉韦谱和长系统发育分支之间高度相似(图)。S7)。这表明过渡突变的共同驱动因素,因此支持由莫努匹拉韦治疗驱动的长系统发育分支。
虽然过渡模式高度相似,但AGILE试验莫努匹拉韦谱包含高速率的G-to-T突变,这在长系统发育分支中不存在(图7,图8)。这种高发生率也存在于安慰剂治疗的患者中,尽管在莫努匹拉韦治疗中该比率似乎更高(图8A)。安慰剂组和莫努匹拉韦组的 G 到 T 突变率似乎高于未经治疗个体的患者突变计算的比率(Tonkin-Hill 等人 (2021),图)。S8),原因尚不清楚。
讨论
我们已经展示了各种证据,这些证据共同表明,莫努匹拉韦治疗的特征在全球测序数据库中是可见的。我们确定了一组长系统发育分支,这些分支表现出大量的过渡突变。这些分支的数量在2022年急剧增加,对于已知暴露于莫努匹拉韦的国家和年龄组来说,它们得到了丰富。这些分支上的突变率升高,与 Fountain-Jones 等人 (2022) 最近对免疫功能低下患者进行的一项研究一致。这些分支表现出与已知接受莫努匹拉韦治疗的患者高度相似的突变谱。测序数据表明,至少在某些情况下,具有大量莫努匹拉韦诱导的替代物的病毒已经传播给其他个体,至少以有限的方式传播。
SARS-CoV-2 的新变体是通过获得增强免疫逃避和内在传播性等特性的突变产生的(Telenti 等人,2022 年;卡拉贝利等人,2023 年)。莫努匹拉韦治疗对变异株生成和传播轨迹的影响难以预测。一方面,莫努匹拉韦增加了宿主中存活病毒群体的序列多样性量,这可能有望提供更多的材料供选择在宿主内向这些增加适应性的特性进化过程中起作用。然而,高比例的诱导突变可能是有害的或中性的,因此有必要考虑与莫努匹拉韦治疗相反的事实。由于莫努匹拉韦导致治疗患者的病毒载量适度降低(Khoo等人,2022),因此在没有治疗的情况下,总病毒载量可能会更高,慢性感染可能会持续更长时间。通过慢性感染产生的变异可能比在莫努匹拉韦治疗期间积累突变的变异更合适,尽管需要更长的时间才能积累相同数量的突变,因此通常来自更古老的而不是当代的谱系。在撰写本文时,我们尚未发现与莫努匹拉韦有关的集群已扩散到超过21人。
我们的工作有一些局限性。检测涉及莫努匹拉韦样特征的特定分支是一种概率判断,而不是绝对判断:莫努匹拉韦只产生少数突变(试验数据表明通常是这种情况),分支长度太小,无法自信地分配突变的原因。因此,我们在这里将分析限制在长分支上。这种方法也可能无法检测到具有大量莫努匹拉韦诱导的突变以及来自其他原因的大量突变(可能发生在慢性感染中)的分支。我们发现不同国家的莫努匹拉韦相关序列的速率差异很大,并表明这部分反映了莫努匹拉韦是否以及如何在不同的地理区域使用——然而,在使用莫努匹拉韦的环境中,基因组测序的速率也会有贡献。例如,如果莫努匹拉韦主要用于老年护理机构,并且这些设施中的病毒比一般社区中的病毒更有可能被测序,这将提高此类序列的确定率。此外,由于作为特定研究的一部分,莫努匹拉韦治疗后的持续检测阳性,可能对一些纳入的序列进行了专门分析。根据不同地点的测序优先级,这种效果可能会有所不同。
我们建议显示这些模式的国家的公共卫生当局进行调查,以确定这些序列或集群是否确实可以直接与莫努匹拉韦的使用联系起来。这些数据将有助于持续评估这种治疗的风险和益处,并可能指导诱变剂作为抗病毒药物的未来发展,特别是对于具有高突变耐受性的病毒,如冠状病毒。
方法
突变注释树的处理
为了识别全球序列数据库中具有莫努匹拉韦相关特征的簇,我们分析了使用 UShER(Turakhia 等人,2021 年)构建的定期更新的突变注释树,其中包含几乎所有全球数据——McBroome 等人 (2021) 树的一个版本。我们使用改编自分类工具的自定义脚本解析树(Sanderson,2022 年)。该脚本将来自排序数据库的元数据添加到每个节点,然后使用简单的启发式方法将这些元数据传递给父节点:(1) 如果父节点的所有后代都用该年份注释了该年份,则父节点被注释为年份,(2) 如果父节点的所有后代都用该国家/地区注释,则父节点使用特定国家/地区进行注释, (3)父节点用其(年龄注释的)后代的平均年龄进行注释。
突变率分析
我们使用 Chronumental (Sanderson, 2021) 为突变注释树中的每个节点分配日期。我们运行了300个步骤的Chrunmental,然后从输出的Newick树中提取了以天为单位的时间长度,并与分支上的突变数量进行比较,根据分支是否满足我们的标准进行拆分,成为高G-to-A分支。
生成聚类树
将排除的发散序列加回突变注释树
我们使用GISAIDR(沃斯和杜兴,2022)从GISAID数据库下载了2022年的所有澳大利亚序列。然后,我们根据序列ID过滤那些在现有突变注释树(MAT)中不存在的那些。我们使用flowalign将这些序列与Hu-1参考对齐。我们修剪了现有的 MAT 以仅保留澳大利亚序列,然后使用 UShER 添加树中缺失的所有序列(Turakhia 等人,2021 年),而不过滤简约的位置或路径长度,以实现澳大利亚的完整 MAT。
长系统发育分支突变谱的计算
使用AGILE试验数据计算莫努匹拉韦和安慰剂突变谱
我们使用先前发表的变异数据计算了莫努匹拉韦和安慰剂SBS光谱(DonovanBanfield等人,2022)。我们使用了第一天(治疗前)和第五天(治疗后)收集的样本的深度测序数据,这些数据来自65名接受安慰剂治疗的患者和58名接受莫努匹拉韦治疗的患者。对于每位患者,我们使用第一天样本的共识序列作为参考序列,并在基因组位点至少5%的读取中将突变确定为偏离患者参考序列的第五天样本中的变异,覆盖率至少为100倍。从患者参考序列中鉴定出每个突变的周围核苷酸背景。
我们将每个突变计数除以武汉-胡-1基因组中起始三胞胎的数量(加入NC_045512.2)和治疗组的患者数量(安慰剂组为65例,莫努匹拉韦组为58例),从而将突变计数转换为突变负担。因此,这通过每个突变在整个基因组中发生的机会数量和每组中的患者数量来重新调整。
为了计算每个突变类别的总突变负担,我们总结了该类别中16个上下文突变的突变负担。通过自举突变计算置信区间。在这里,治疗中的原始突变集在通过三胞胎可用性和患者数量重新缩放之前重新采样。运行 1000 个引导程序,计算 95% 置信区间。
为了能够与包含宿主感染期间获得的突变的其他 SARS-CoV-2 数据集进行比较,我们从之前的一项深度测序研究 Tonkin-Hill 等人(2021 年)中获得了变异。我们使用武汉-胡-1基因组确定了每个突变的周围核苷酸背景,并根据上下文可用性进行了重新缩放。
突变谱的比较
安慰剂和莫努匹拉韦光谱之间的余弦相似性是使用MutTui(https://github.com/chrisruis/MutTui)计算的。
我们通过回归每种上下文中突变类中突变的比例,比较了长系统发育分支和AGILE莫努匹拉韦光谱之间每个过渡突变内的上下文模式。为了评估相关性的显著性,我们随机化了每个谱中突变类别内的比例,并重新计算了相关性。进行了 1000 次随机化,p 值计算为相关性至少与真实数据一样大的随机化比例。
数据可用性
https://github.com/theosanderson/molnupiravir
数据可用性
本研究的结果基于截至 14 年 449 月与 GISAID 上可用的 737,2022,10 个序列相关的元数据,可通过 55876.8/gis230110.10wz 和 55876.8/gis230110.6db 访问(另见补充表)。这项研究的结果也基于INSDC的594,478,<>个序列 - 作者,元数据和序列可在此处获得。在构建突变注释树之前,对两个数据库中存在的数据进行重复数据删除。
作者贡献
RH确定了初始分支,以及它们与莫努匹拉韦的可能联系。TS对突变注释的树和全局元数据进行了分析。CR进行了所有突变光谱分析。ID-B为AGILE试验数据创建了生物信息学管道。所有作者都参与了文稿写作。
资金
TS得到了威康信托基金会(210918/Z/18/Z)和弗朗西斯·克里克研究所的支持,该研究所的核心资金来自英国癌症研究中心(FC001043),英国医学研究委员会(FC001043)和威康信托基金会(FC001043)。这项研究全部或部分由威康信托基金会资助[210918/Z/18/Z,FC001043]。出于开放获取的目的,作者已对本预印本产生的任何作者接受的手稿应用了CC-BY公共版权许可。
ID-B得到了利物浦大学国家卫生与护理研究所(NIHR)新兴和人畜共患感染健康保护研究部(HPRU)的博士资助,与英格兰公共卫生(PHE)(现为UKHSA)合作,与利物浦热带医学院和牛津大学合作(200907)。所表达的观点是作者的观点,不一定是卫生和社会护理部或NIHR的观点。资助者或试验申办者都没有参与研究设计、数据收集、分析、解释或手稿的准备。
TP由MRC资助的G2P-UK国家病毒学联盟资助(MR / W005611 / 1)。
CR was supported by a Fondation Botnar Research Award (Programme grant 6063) and UK Cystic Fibrosis Trust (Innovation Hub Award 001).
Supplementary Information
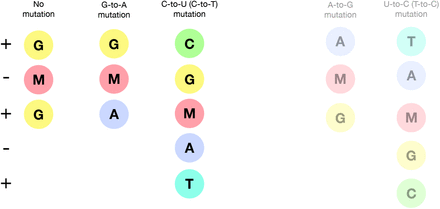
Figure S1.Possible outcomes from MTP incorporation
该图描述了与MTP并入MTP相关的一些突变途径。第一列显示可能是常见事件,但无法通过排序检测到。MTP可以掺入RNA(与G配对),然后在下一轮合成中再次与G配对,这将导致最终序列中没有突变。然而,如果MTP在掺入后呈现出另一种互变异构形式,它可以与A结合,产生G到A突变。第三列显示,如果正义碱基是C,那么这将在负义基因组的形成中与G结合。在随后的复制中,这种负义基因组可以经历与第二列相同的G-to-A突变,最终导致正义C-to-T突变。尽管互变异构体形式对游离和掺入的MTP核苷酸的偏向似乎有利于突变的这些方向性,但反过来也是可能的,导致A--G和T--C突变。
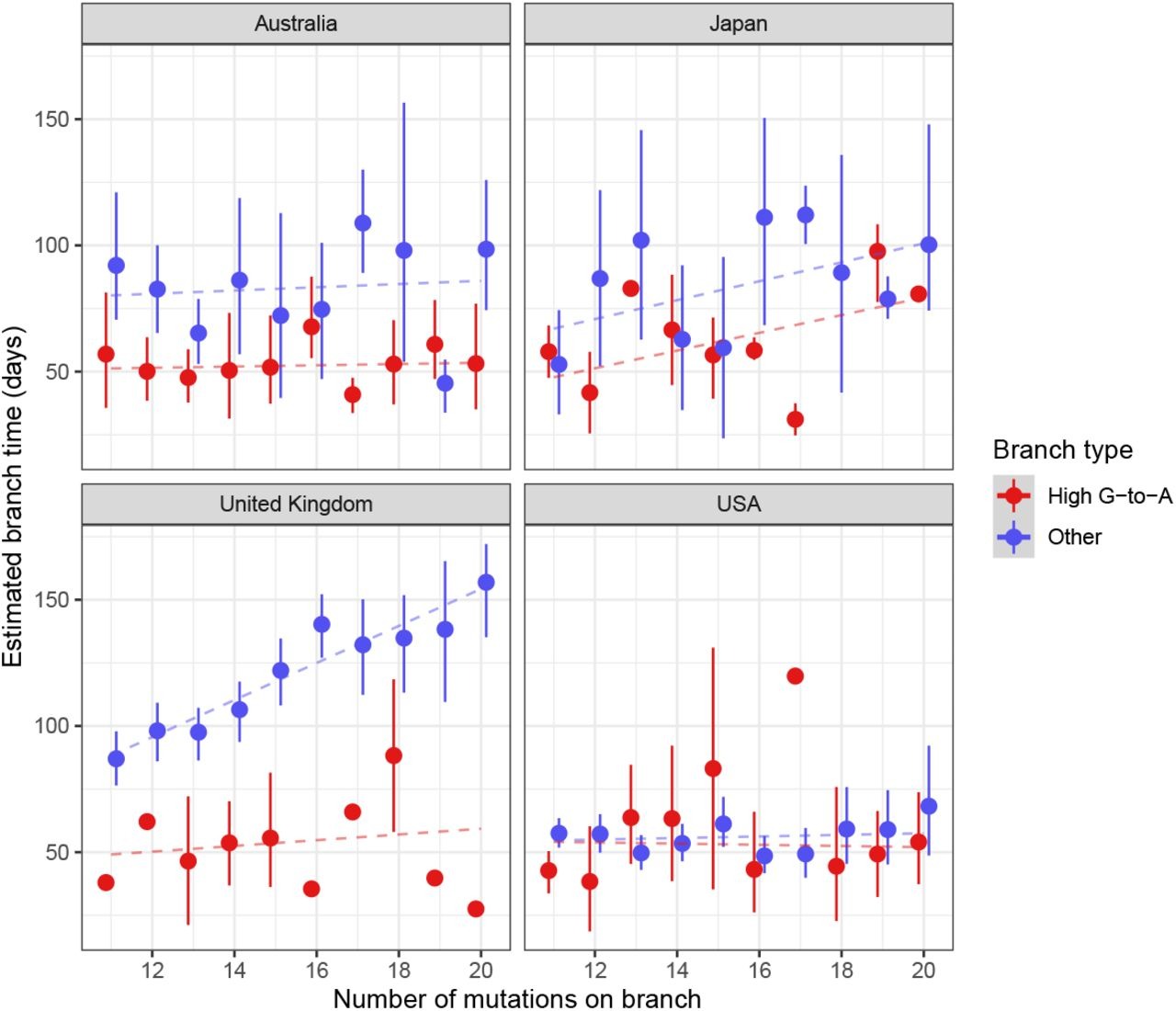
图 S2.高G-to-A分支涉及比其他分支类型在更短的时间内发生的相同数量的突变
我们使用Chronumental为全局UShER突变注释树中的所有节点分配日期,然后分离分支长度>10和<= 20,并绘制了平均时间长度及其95%置信区间突变数,以及拟合的线性模型,用于澳大利亚,日本,英国和美国。这种分析应该只被视为半定量的,因为该算法将试图将其时间顺序与整体SARS-CoV-2突变率相协调,因此是保守的,并且测序错误和重组也可以在短时间内驱动大量的明显突变率。

图 S3.来自澳大利亚的高 G 到 A 分支在降采样的全球树上可视化
从澳大利亚序列中制作了完整的MAT,并鉴定了来自高G-to-A事件的基因组。然后使用Usher.bio创建一个树,这些序列在下采样的全局树上突出显示,并使用Nextstrain进行可视化。

图 S4.AGILE 试验数据集中安慰剂和莫努匹拉韦治疗之间主要 SARS-CoV-2 变异的分布。
显示了感染每种变体的患者比例。比例相似,表明安慰剂和莫努匹拉韦光谱之间的差异不会受到先前观察到的变体之间光谱差异的影响(Ruis等人,Bloom等人)。VOC = 关注的变体。

Figure S5.突变谱中的上下文位置。
RNA突变谱包含12种突变类型,例如C-to-T。该光谱还捕获每个突变周围的核苷酸。有四个潜在的上游核苷酸和四个潜在的下游核苷酸。此图显示了示例突变类型中 16 个上下文中每个上下文的位置。例如,最左边的条形表示 ACA 上下文中的 C 到 T 突变,而最左边的第二个条形表示 ACC 上下文中的 C 到 T 突变。
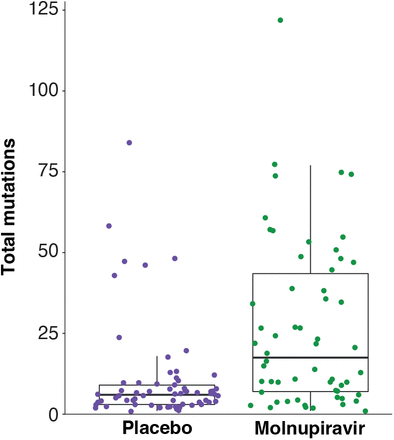
图 S6.安慰剂治疗患者和莫努匹拉韦治疗患者的突变负荷比较。
所有替代类别的突变总数绘制在 AGILE 试验中每位患者的第 5 天样本中。
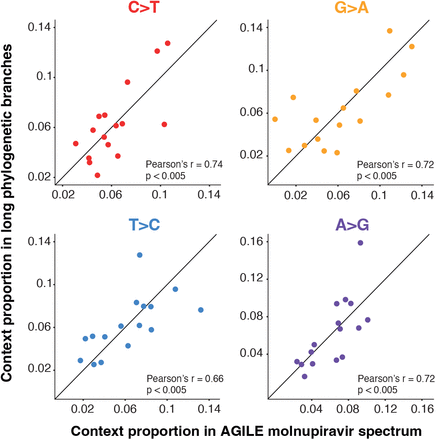
图 S7.比较根据AGILE试验数据计算的莫努匹拉韦谱与长系统发育分支谱之间的过渡突变中的上下文模式。
显示了相应转换中突变的比例(例如,ACA环境中的C>T突变的比例)。P 值表示上下文随机化的比例,皮尔逊 r 相关性至少与真实数据一样大(参见方法)。比例根据上下文在基因组中出现的次数进行归一化。
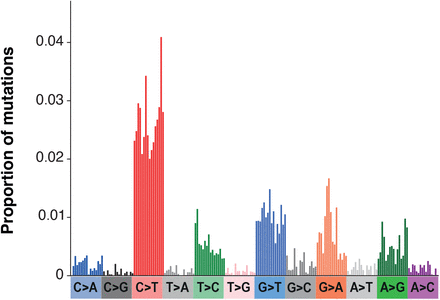
图 S8.在未经治疗的患者中获得的突变谱。
确认
我们非常感谢所有数据贡献者,即负责获取标本的作者及其原始实验室,以及他们的提交实验室,用于生成基因序列和元数据并通过GISAID倡议进行共享,这是本研究的基础。我们也非常感谢所有为生成基因组做出贡献的人,这些基因组已存放在INSDC数据库中,这项研究也是基于该数据库。我们感谢Angie Hinrichs及其同事访问了使用所有可用基因组数据构建的UShER突变注释树。我们感谢Jesse Bloom,Michael Lin,Richard Neher和Kelley Harris的有益讨论。此预印本使用Stephen Royle和Ricardo Henriques的LaTeX模板。