Гемолитические анемии: ферментопатии и мембранопатии
https://t.me/medach
Мы продолжаем рассматривать гемолитические анемии, обусловленные внутриклеточным гемолизом, при котором эритроциты разрушаются макрофагами, гранулоцитами, NK-клетками в селезенке, печени и костном мозге.
Напомним, что причинам патологического внутриклеточного гемолиза относятся:
- Наследственные нарушения синтеза или структуры гемоглобина (гемоглобинопатии);
- Наследственные нарушения активности ферментов эритроцитов (ферментопатии);
- Наследственные нарушения структуры мембраны эритроцитов (мембранопатии);
- Несовместимость по антигенам системы АВО или резус (Rh) между матерью и плодом, при гемотрансфузиях;
- Избыточное количество эритроцитов (физиологическая желтуха, эритробластоз новорожденного, эритремия — при количестве эритроцитов более 6-7 х 10^12/л).
Гемоглобинопатии (серповидно-клеточную анемию и талассемию) мы рассмотрели ранее, сейчас же речь пойдет об ферментопатиях и мембранопатиях.
В настоящее время известно более 20 наследственных ферментопатий эритроцитов, приводящих к потере устойчивости клеток к воздействиям различных окислителей (лекарственных препаратов и др.), укорочению продолжительности их жизни и усиленному гемолизу. Чаще всего встречаются ферментопатии, связанные с дефицитом глюкозо-6-фосфатдегидрогеназы (Г-6-ФДГ, фермент пентозофосфатного шунта, через который метаболизируется около 10 % глюкозы) и пируваткиназы (фермент гликолиза).
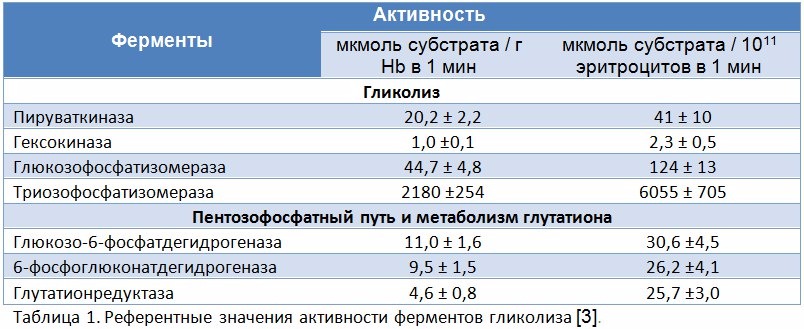
Лабораторное подтверждение эритроцитарных ферментопатий основано на биохимическом определении активности ферментов в гемолизате (таблица № 1) [1, 3].
Гемолитическая анемия, обусловленная недостаточностью Г-6-ФДГ — самая распространенная эритроцитарная ферментопатия (частота встречаемости 1:20 - 1:40) [3, 9]. К началу 2018 года выявлено более 200 мутаций в гене G6PD, отвечающем за синтез фермента и расположенном на Х-хромосоме [8]. Все эти мутации приводят к снижению активности фермента в различной степени; полное отсутствие активности Г-6-ФДГ летально. Наследование недостаточности Г-6-ФДГ является Х-сцепленным, поэтому значительно чаще возникает у мужчин [1, 3, 8, 9]. Однако, вопреки распространённому мнению, это заболевание не рецессивно, поскольку проявляется и у женщин-гетерозигот [8]. У таких женщин может наблюдаться как полное отсутствие активности Г-6-ФДГ, так и её нормальный уровень. Объясняется это случайной инактивацией одной из двух Х-хромосом, происходящей у эмбрионов женского пола [1, 3, 6, 8, 9].
Недостаточность Г-6-ФДГ чаще встречается у жителей или выходцев из стран «малярийного пояса» (Африки, Юго-Восточной Азии, Средней Азии, Средиземноморья и Закавказья) [3, 4, 6]. Показано, что некоторые мутации гена G6PD придают эритроцитам резистентность к малярийным плазмодиям Plasmodium falciparum и Plasmodium vivax [7, 9]. В Греции и на Ближнем Востоке недостаточность Г-6-ФДГ присутствует у 35–40 % мужчин [6]; среди русских распространённость ферментопатии составляет до 2 % [3].
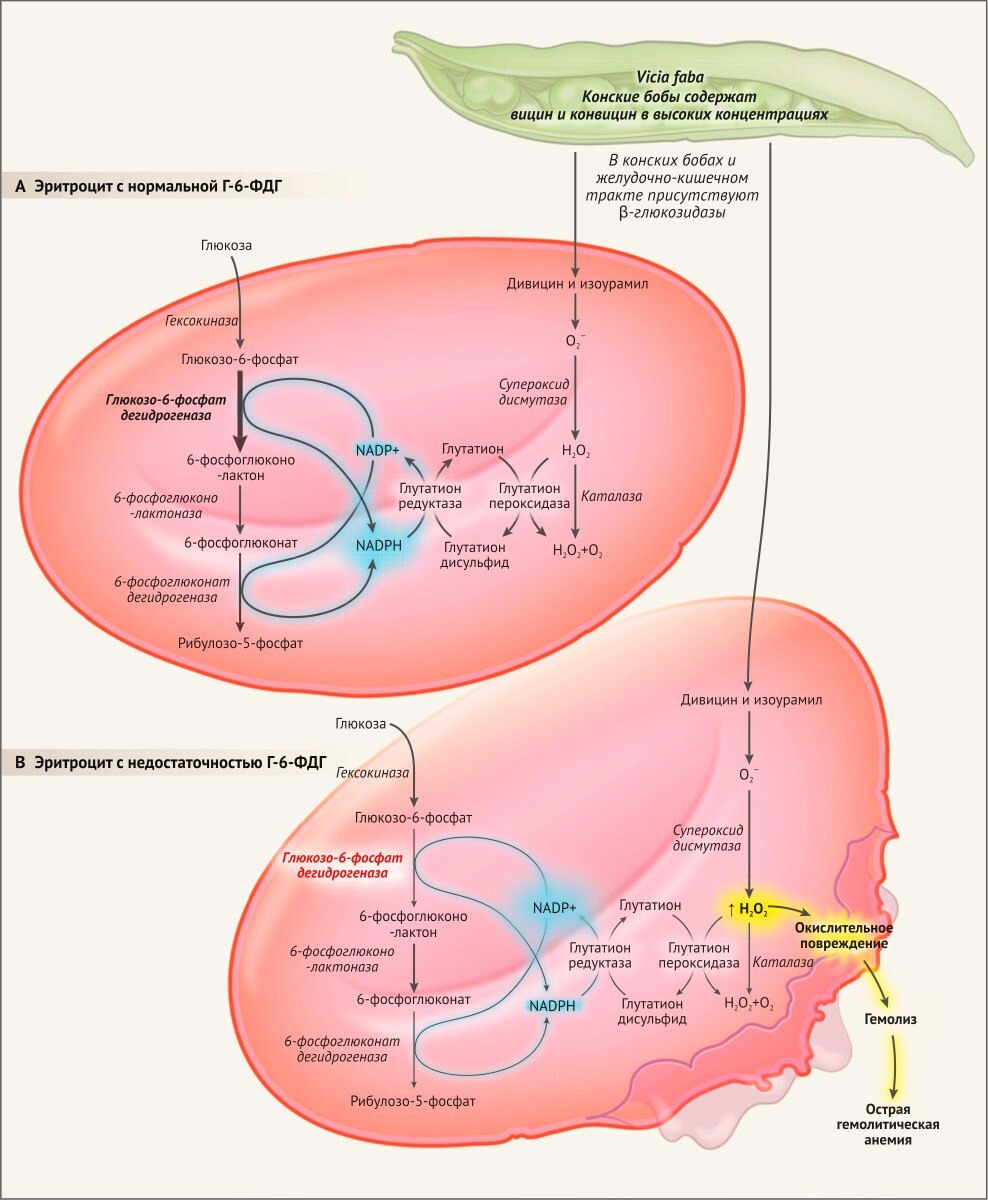
Срок жизни Г-6-ФДГ-дефицитных эритроцитов не превышает 30 дней, они очень быстро стареют и гибнут. При недостаточной активности Г-6-ФДГ нарушается пентозофосфатный путь, и не происходит образования восстановленного глютатиона, который защищает гемоглобин и мембрану эритроцитов от разного рода окислителей. C истощением запасов восстановленного глютатиона в эритроците резко усиливаются окислительные процессы, приводящие к инактивации внутриклеточных ферментных систем и повышению проницаемости мембраны эритроцита, что ведет к его набуханию и гемолизу (рис. 1). Следует отметить, что при недостаточности Г-6-ФДГ помимо внутриклеточного гемолиза развивается и внутрисосудистый гемолиз [1, 4, 7].
Выделяют 4 клинические формы проявления недостаточности Г-6-ФДГ [7]:
1) острая гемолитическая анемия;
2) хроническая гемолитическая анемия;
3) гипербилирубинемия новорожденных;
4) фавизм.
Острую гемолитическую анемию при недостаточности Г-6-ФДГ могут спровоцировать инфекционные заболевания (точнее, эндогенные окислители, вырабатываемые нейтрофилами в ответ на инфекцию [5]), контакт с нафталином [6, 9], диабетический ацидоз [1] или же приём некоторых лекарственных препаратов, чаще всего хинолинового ряда, сульфаниламидов, анальгетиков, витамина К [4]. Клинические симптомы могут возникать внезапно через несколько часов после приема препарата или же развиваться постепенно в течение 2-3 суток [3, 9]. Первыми симптомами обычно бывают иктеричность склер и потемнение мочи. Если контакт с провоцирующим гемолиз агентом не прекращается, на 4-5 сутки возникает гемолитический криз с выделением тёмной мочи. При тяжёлом течении болезни повышается температура, появляется головная боль, рвота, иногда понос. Возникает одышка, часто увеличивается селезенка [3]. При молниеносных формах заболевания наступает смерть от шока или острой аноксии [1]. Существует феномен, так называемого самоограничения гемолиза, когда в самый разгар клинических проявлений гемолитического криза внутрисосудистый гемолиз внезапно прекращается. Это связано с преимущественной гибелью старых и дефектных эритроцитов [1].
Хроническая гемолитическая анемия протекает легче острой формы. Больные жалуются на постоянную иктеричность склер, периодическое усиление желтушности кожи. Выявляется увеличение селезёнки, анемия в отсутствие инфекции или известных провоцирующих факторов [1, 7].
Гипербилирубинемия новорожденных при недостаточности Г-6-ФДГ возникает редко. Проявляется это состояние на 2-3 сутки после рождения анемией и желтушностью. Возможно развитие ядерной желтухи (билирубиновой энцефалопатии) и необратимого поражения мозга [7, 9].
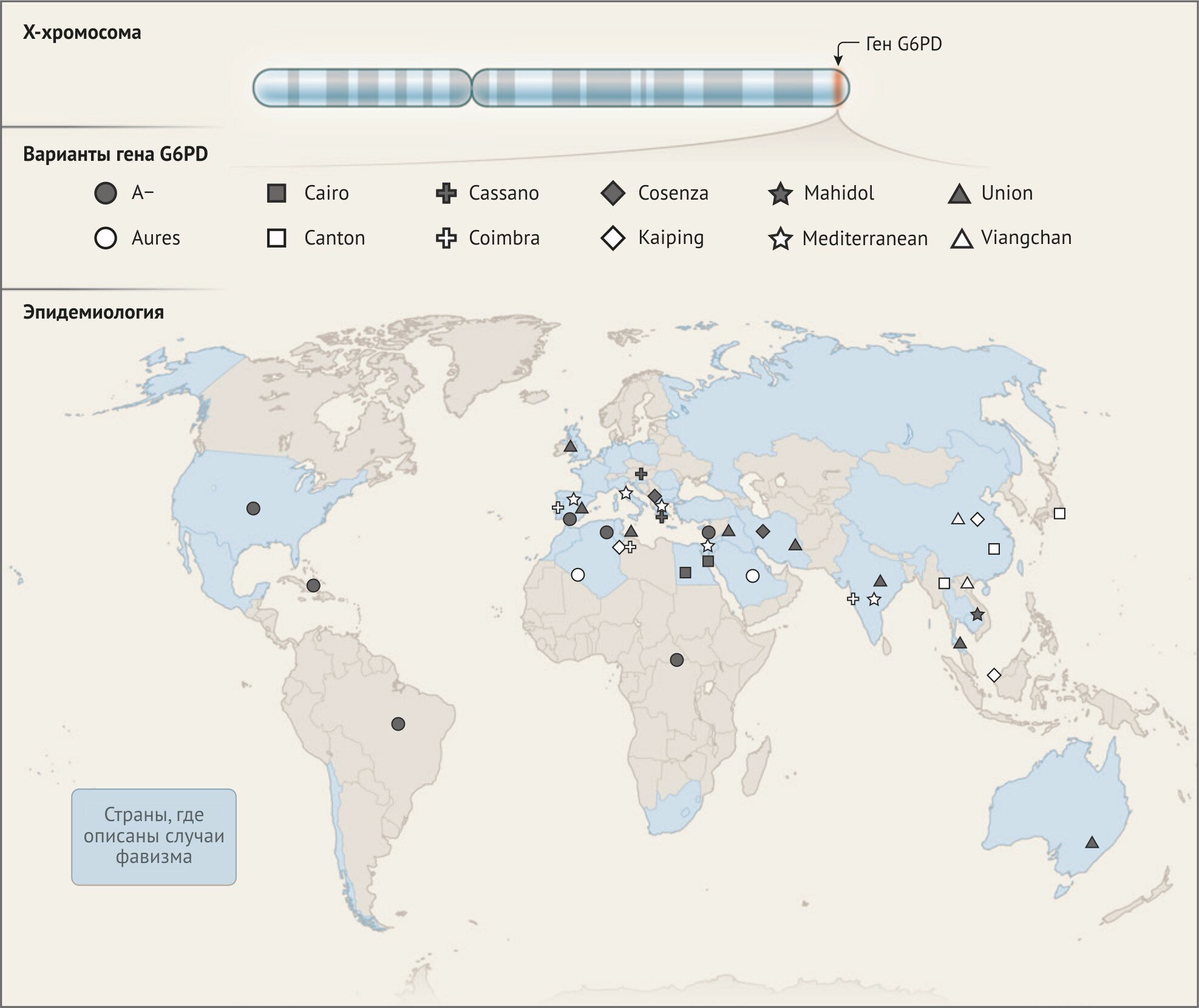
Фавизм – частный и наиболее распространённый случай Г-6-ФДГ-дефицитной анемии (частота 1:10 000-1:50 000) [1, 4, 8]. В этом случае гемолиз возникает после употребления в пищу бобов (англ. favа beans), которые высвобождают оксиданты при переваривании в желудочно-кишечном тракте [7]. Особенностью фавизма является острый гемолиз, наступающий быстрее, чем в случае приёма лекарств, но протекающий легче и сопровождающийся диспепсией и болью в животе [3, 8]. Распространено заблуждение, что фавизм может возникать при вдыхании цветочной пыльцы. Однако ни один случай развития именно гемолитической анемии (в отличие от аллергических реакций) при контакте с пыльцой документально не подтверждён [8]. Фавизм встречается в Италии, Греции, Турции, Болгарии, Ираке, Китае, Америке, Азербайджане [4]. Причиной фавизма являются по меньшей мере 14 различных мутаций в гене Г-6-ФДГ (рис. 2) [8].
Лабораторные показатели при недостаточности глюкозо-6 фосфатдегидрогеназы и фавизме [1, 3, 5-8]:
• В крови — анемия. Уровень гемоглобина снижается до 60 г/л, в период криза — до 20 г/л. Также снижается число эритроцитов (до 1 х 10^12/л) и гематокрит. Повышаются показатели MCV и МСН. Содержание ретикулоцитов увеличивается до 20-25 % и зависит от степени гемолиза. Выявляется умеренный лейкоцитоз со сдвигом до миелоцитов. Количество тромбоцитов обычно не меняется.
• В мазке крови анизоцитоз, пойкилоцитоз, полихроматофилия, базофильная пунктация, тельца Жолли. При тяжелом гемолитическом кризе может выявляться большое количество телец Гейнца (рис. 3).
• В сыворотке крови повышается содержание свободного гемоглобина (внутрисосудистый гемолиз) и железа, часто увеличивается концентрация непрямого билирубина. Уровень гаптоглобина снижается вплоть до полного отсутствия.
• В моче гемоглобинурия (до 2–3 г/л), гемосидеринурия.
• Кал темный, повышен стеркобилиноген.
Диагностика основана на определении уровня фермента Г-6-ФДГ спектрофотометрическим методом.
Лечение больных с гемолитическим кризом начинают с устранения влияния окислителей (например, отмены препаратов, вызывающих гемолиз) и назначения антиоксидантной терапии. Проводят мероприятия противошокового характера [1]. При тяжёлой анемии (Hb <70 г/л или Hb <90 г/л в сочетании с гемоглобинурией) необходимы гемотрансфузии. В лёгких случаях достаточно регидратации и симптоматического лечения [8]. Прогноз при острых гемолитических кризах зависит от скорости устранения лекарственного препарата или вещества, вызвавшего криз, возраста и общего состояния больного. При развитии анурии и почечной недостаточности прогноз неблагоприятен. Возможно ухудшение заболевания до апластического состояния или миелодиспластического синдрома, миелофиброза. В случае благоприятного исхода криза наступает выздоровление с нормализацией картины крови. Однако могут периодически появляться симптомы минимального гемолиза и признаки легкой желтушности [1].
Дефицит пируваткиназы — вторая по частоте ферментопатия после дефицита Г-6-ФДГ (частота 1:20 000). Наследуется по аутосомно-рецессивному типу и встречается во всех этнических группах [1]. Пируваткиназа является одним из основных ферментов гликолиза. Её функция заключается в образовании АТФ на заключительном этапе гликолиза (рис. 4) [1, 3].
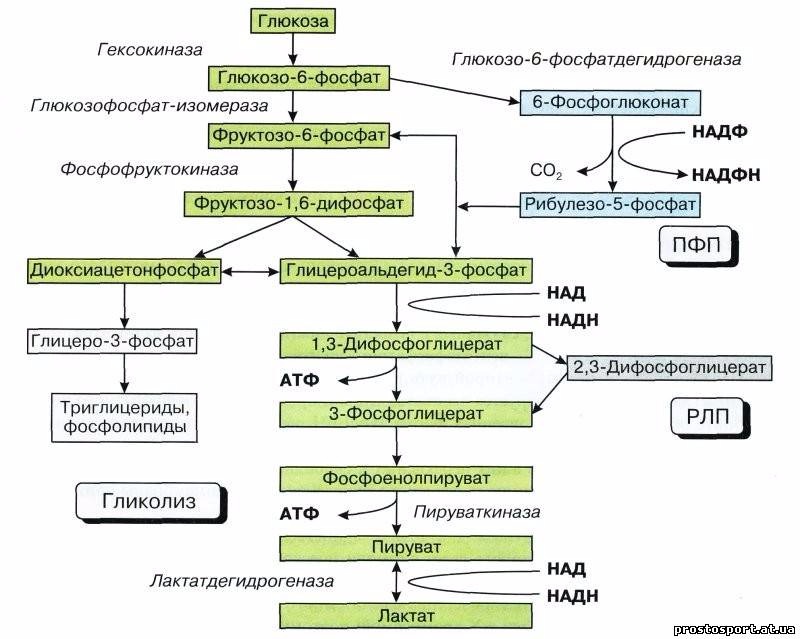
Недостаточность пируваткиназы возникает из-за мутаций в гене PKLR. На данный момент описано около 180 мутаций. Заболевание наследуется по аутосомно-рецессивному типу [9].
При дефиците пируваткиназы в эритроцитах накапливаются промежуточные продукты гликолиза и снижается содержание АТФ. Это сопровождается нарушением функции АТФ-азного насоса эритроцита и потерей ионов калия, что провоцирует гемолиз эритроцитов. Клинические проявления дефицита пируваткиназы очень разнообразны, от отсутствия каких-либо признаков заболевания до тяжёлой анемии, требующей постоянных переливаний крови. Чаще заболевание проявляется в первые годы жизни ребенка; возникает бледность кожных покровов, желтуха, спленомегалия [1, 3, 6, 9].
Лабораторные показатели при дефиците пируваткиназы [3]:
• В крови — нормохромная несфероцитарная анемия, эритроцитарные индексы близки к норме. Возможно снижение гемоглобина (до 40 г/л) и эритроцитов. Количество лейкоцитов и тромбоцитов обычно нормальное.
• В мазке — анизоцитоз и пойкилоцитоз, может наблюдаться полихроматофилия и эритроциты с базофильной пунктацией, иногда мишеневидные эритроциты, эритрокариоциты (рис. 5). Ретикулоцитоз в период криза может достигать 70 %.
• В сыворотке крови при гемолитическом кризе повышен непрямой билирубин.
• В костном мозге выраженный эритрокариоцитоз.
Главным критерием диагностики выступает снижение активности пируваткиназы ниже 30 % от нормы. Возможно молекулярно-генетическое тестирование. Лечение дефицита пируваткиназы проводится в случае тяжёлой анемии и заключается в переливании эритроцитарной массы для поддержания уровня гемоглобина >70 г/л [1].
Дифференциальная диагностика недостаточности пируваткиназы, как и недостаточности Г-6-ФДГ, проводится с другими, более редкими эритроцитарными ферментопатиями (таблица № 2) [6]. Ещё один тип анемий с внутриклеточным гемолизом — мембранопатии, причиной которых является генетический дефект структурных белков эритроцитов (таблица № 3).
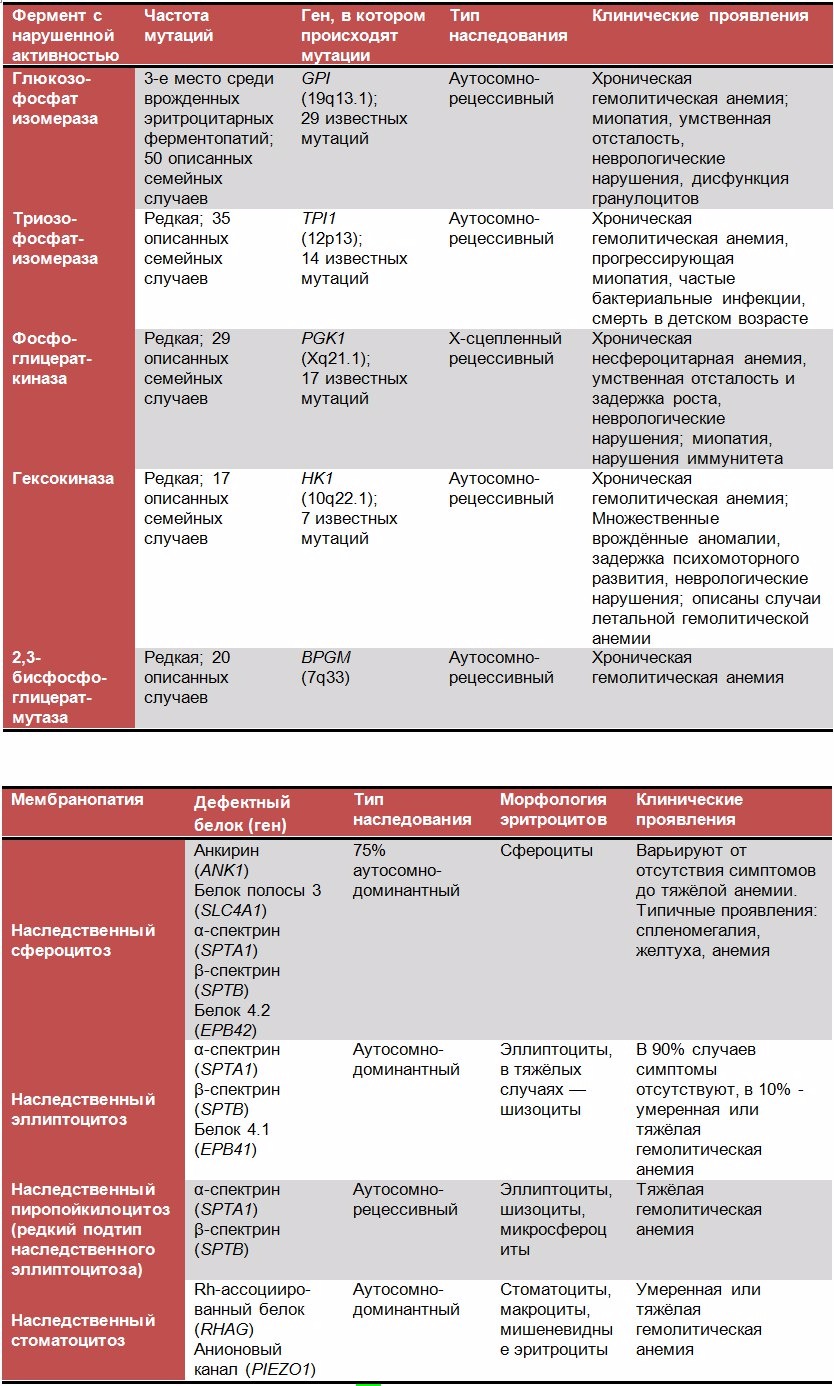
Таблица 3. Мембранопатии эритроцитов.
К таким мембранопатиям относится наследственная микросфероцитарная анемия Минковского – Шоффара. Это заболевание очень подробно описано в двух постах:
Поэтому здесь мы его рассматривать не будем, а обратим внимание на овалоцитоз и стоматоцитоз.
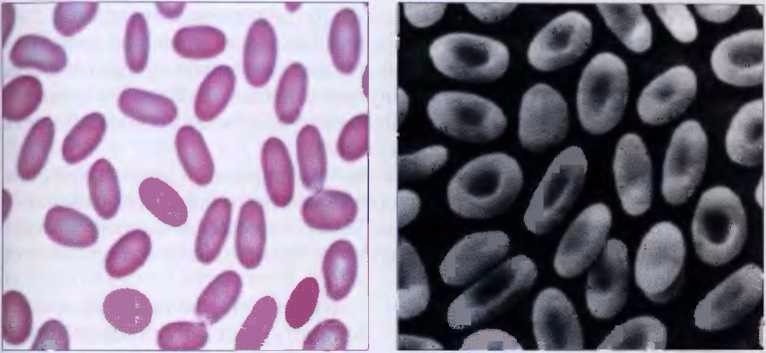
Овалоцитарная гемолитическая анемия (наследственный овалоцитоз, эллиптоцитоз) — это редкое заболевание, которое встречается во многих этнических группах и наследуется по аутосомно-доминантному типу [3, 5]. Возникает овалоцитоз из-за ослабления связей между молекулами белка спектрина или дефекта комплекса спектрин—актин—протеин 4.1R. Наиболее частой причиной (65 % случаев) наследственного овалоцитоза является мутация α-спектрина.
В большинстве случаев овалоцитоз не имеет клинических проявлений, однако примерно у 10 % обладателей мутации развивается анемия средней тяжести, очень схожая с микросфероцитозом. Заболевание характеризуется хроническим нетяжелым течением с гемолитическими кризами, желтухой и анемией, спленомегалией, изменениями скелета, в частности черепа; возможны трофические язвы голени [3].
Лабораторные показатели при наследственном овалоцитозе [3, 5]:
• В крови — нормохромная несфероцитарная анемия с высоким ретикулоцитозом. Объем эритроцитов (MCV) и концентрация гемоглобина (МСН) в норме. Показатель RDW повышен. Осмотическая резистентность эритроцитов понижена, СОЭ ускорена.
• В мазке — овалоциты, до 40-50 % при гетерозиготном носительстве и до 96 % при гомозиготном состоянии (рис. 6).
• В костном мозге регенераторный или гиперрегенераторный тип кроветворения с преобладанием эритробластов.
Стоматоцитарная гемолитическая анемия (стоматоцитоз) — редкое заболевание с аутосомно-доминантным типом наследования [3, 9]. В основе заболевания лежит изменение функции структурных белков мембраны эритроцитов, приводящее к нарушению регуляции объема клетки. Клинические проявления различны —от бессимптомного носительства до тяжёлой гемолитической анемии, напоминающей микросфероцитоз. Внутриклеточный гемолиз эритроцитов сопровождается увеличением селезенки, желтухой. Возможны изменения скелета [3].
Лабораторные показатели при стоматоцитозе [3, 9]:
• В крови — нормохромная несфероцитарная анемия с высоким ретикулоцитозом. Объем эритроцитов (MCV) и концентрация гемоглобина (МСН) в норме. Осмотическая резистентность эритроцитов может быть понижена, СОЭ ускорена.
• Особенность болезни - стоматоцитоз (рис. 7), который характеризуется наличием в центре клетки неокрашенного участка в виде вытянутой светлой полосы, напоминающей форму рта (stoma - рот).
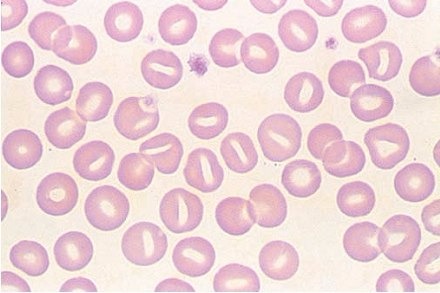
• В сыворотке крови повышен непрямой билирубин.
• Костный мозг гиперклеточный, выражен эритрокариоцитоз.
Источники:
- Соколова Т. А. Генетические аспекты наследственных гемолитических анемий (энзимопатий) //Успехи современного естествознания. – 2012. – №. 10.
- Соколова Т. А. Генетические аспекты наследственных гемолитических анемий (мембранопатий) //Успехи современного естествознания. – 2012. – №. 10.
- Долгов В. В. Лабораторная диагностика анемий. – 2009.
- Иванов В.А., Манзурова Е.Н. Некоторые заболевания системы крови. Анемии. – Учебно-методическое пособие. –Курск. – 2015.
- Bain B. J. A beginner's guide to blood cells. – John Wiley & Sons, 2017.
- Bain B. J. Blood cells: a practical guide. – John Wiley & Sons, 2014.
- Nayak R., Rai S., Gupta A. Essentials in hematology and clinical pathology. – JP Medical Ltd, 2011.
- Luzzatto L., Arese P. Favism and Glucose-6-phosphate Dehydrogenase Deficiency //New England Journal of Medicine. – 2018. – Т. 378. – №. 1. – С. 60-71.
- Keohane E. M., Smith L. J., Walenga J. M. Rodak's hematology: clinical principles and applications. – Elsevier Health Sciences, 2015.