Пациенты с редкими заболеваниями
https://t.me/medach
Болезнь «хрустального человека», прогерия, лизосомные болезни накопления, боковой амиотрофический склероз – эти и еще около 8000 известных на сегодняшний день болезней относятся к группе так называемых редких заболеваний. Приблизительно 80% таких заболеваний являются генетически обусловленными. Также среди недугов данной группы можно встретить инфекционные и аутоиммунные болезни. Для некоторых из них причина развития пока не установлена наверняка, и уж совсем не многие из редких заболеваний излечимы. Помимо редкой встречаемости среди населения, редкие болезни объединяет то, что, по большей части, они манифестируют в детском возрасте, обладают хроническим течением, сопровождаются инвалидизацией и ограничивают продолжительность жизни пациента.
Редкость отдельных заболеваний представляет по медицинским и экономическим основаниям немалую трудность как для медицинского обслуживания пациентов, так и для проведения исследований, направленных на обеспечение более достоверной диагностики и эффективного терапевтического воздействия. Таким образом, по причине малого количества пациентов редкие заболевания зачастую не распознаются врачами, а потому лечатся либо неверно, либо вовсе не лечатся. Соответственно, частыми последствиями диагностических ошибок являются неадекватное назначение медикаментов или ненужное оперативное вмешательство, ведущие к неминуемым осложнениям. По этой причине пациенты с диагностированными редкими заболеваниями, а также пациенты с неясным диагнозом должны в обязательном порядке осматриваться целой группой узких специалистов, причем желательно, чтобы эти специалисты были доступны в пределах одного специализированного центра. Совместная обработка данных о пациенте врачами разных специальностей может дать более плодотворные результаты и стать действительно эффективно работающим методом помощи больным с редкими заболеваниями. Ведь, чаще всего, того пациента, диагноз которого тот или иной врач распознать не в состоянии, просто отправляют от одного доктора к другому. Такая эпопея оканчивается безвозвратной потерей времени, которое невероятно важно для оказания своевременной помощи. Хотя, безусловно, для налаживания работы подобных центров необходим запуск немалой по объему реорганизации работы медицинских учреждений.
Рассмотрим несколько примеров трудностей диагностики редких заболеваний, с которыми врачи могут столкнуться. Лизосомные болезни накопления в педиатрической практике довольно часто упускаются, могут быть неверно определены или же распознаны слишком поздно, вследствие чего шансы на успешную терапию неумолимо снижаются. К лизосомным болезням с частотой встречаемости 1:7500 относятся болезнь Фабри, болезнь Гоше, болезнь Хантера. Это системные заболевания с широким разнообразием зачастую неспецифической (а потому трудно классифицируемой) симптоматики. Врожденные заболевания обмена веществ могут проявиться в любом возрасте, в 50% случаев впервые болезнь манифестирует на первом году жизни. Однако примером того, как важно перепроверять предположения в случае затруднительной диагностики патологий обмена веществ, является случай одного 15-тилетнего мальчика. Ребенок существенно отставал в росте и с 4-х лет получал гормональную терапию СТГ. Мальчик посещал школу, где имел хорошую успеваемость, а также три раза в неделю занимался спортом. Он обратился к врачу с жалобами на рецидивирующие боли в нижних конечностях и точечные красноватые образования на обоих коленях (ангиокератомы).
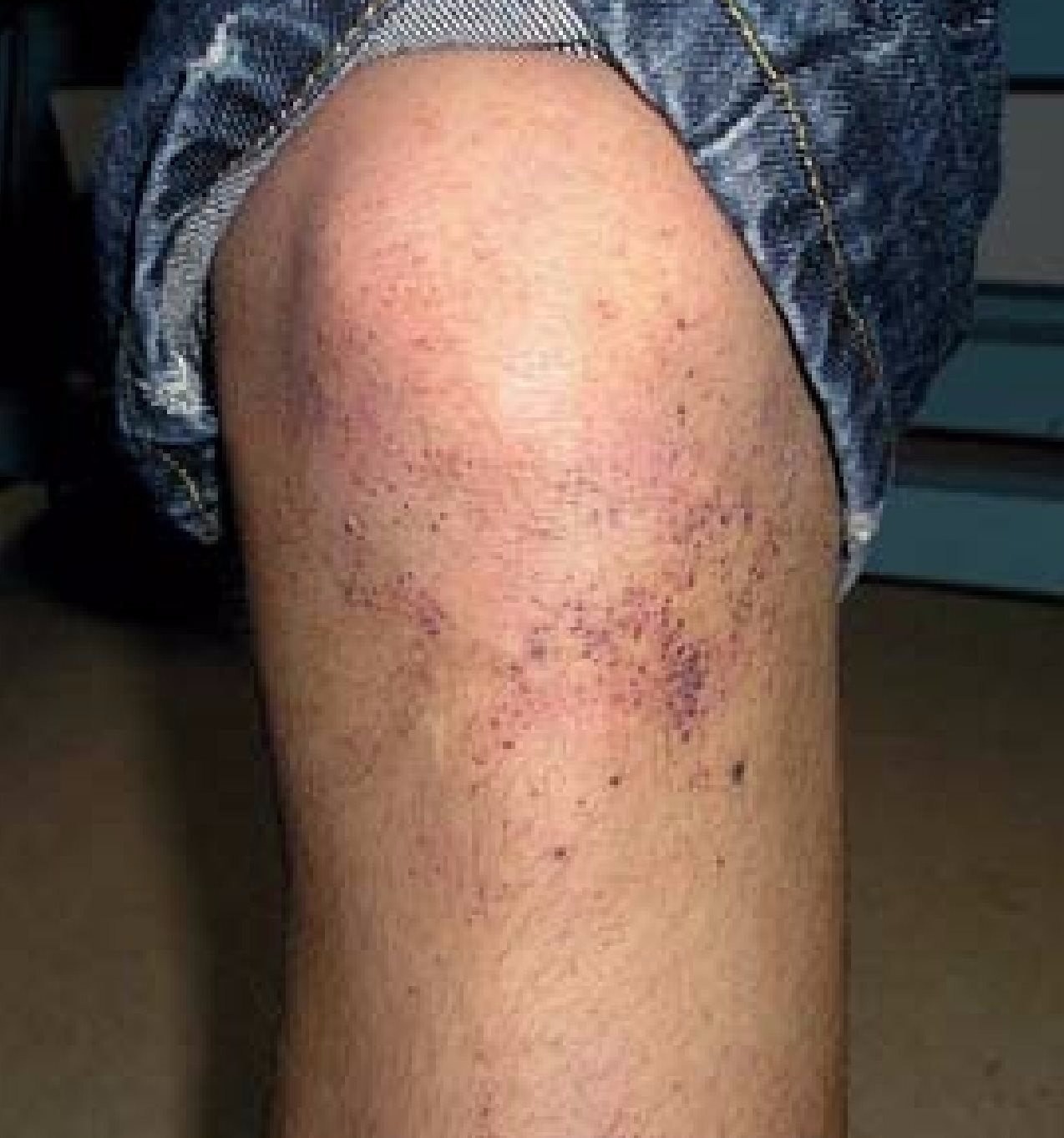
Клиническое обследование и результаты лабораторных анализов не дали поначалу никакой информации. При внимательном осмотре лица были обнаружены ангиокератомы по краю губ.

При дальнейших исследованиях была выявлена легкая протеинурия. УЗИ брюшной полости и рентген грудной клетки без изменений. Ведущим к верной постановке диагноза в данном случае были ангиокератомы. У детей с болезнью Фабри как инициальные симптомы в 80% случав наблюдаются ангиокератомы, акропарестезии, а также нечувствительность к теплу/холоду, гипогидроз. Заболевание относится к сцепленным с Х-хромосомой, могут заболевать как девочки, так и мальчики. Наследуются мутации гена альфа-галактозидазы (GLA), которые становятся причиной того, что фермент альфа-галактозидаза синтезируется в недостаточном для лизосомного разрушения глоботриаозилцерамида (Gb3) количестве, либо не синтезируется вовсе, поэтому Gb3 аккумулируется в клетках. Это ведет к прогрессирующему нарушению клеточных функций и полиорганному повреждению. Дети часто страдают от приступообразных нейропатических болей и имеют заболевания ЖКТ в анамнезе. Однако единой картины классической симптоматики нет, бывает, что встречается только какой-либо один симптом, либо их произвольное сочетание, в чем и заключается сложность идентификации конкретной лизосомной болезни.
Для уточнения диагноза пациентам проводят проверку активности альфа-галактозидазы, иногда (в особенности пациенткам) рекомендуется проведение молекулярно-генетического исследования. Еще одним дополнительным признаком, свидетельствующим о наличии болезни Фабри, является обнаружение специфического помутнения роговицы (сornea verticillata). Прогрессия заболевания может быть сдержана посредством ферментотерапии (например, агалсидаза альфа). Более подробно о болезни Фабри можно прочитать в нашем посте, вышедшем ранее.
В качестве второго примера поздно диагностированной болезни накопления возьмем случай одной 10-летней девочки. В течение пяти недель ее беспокоили сильные боли и припухлость правой голени. Температуры не было, время от времени случались носовые кровотечения. За 6 месяцев до описанных симптомов на осмотре врач пальпаторно определил увеличенные размеры печени. По результатам анализа крови была отмечена тромбоцито- и лейкопения, гемоглобин был несколько снижен. УЗ исследование мягких тканей голени и брюшной полости позволило обнаружить остеонекроз и гепатоспленомегалию.
У девочки была болезнь Гоше – одна из наиболее часто встречаемых из около полусотни лизосомных болезней накопления. Для подтверждения диагноза рекомендуется определение уровня активности хитотриозидазы и бета-глюкоцереброзидазы в крови.
Обширное разнообразие симптомов при болезни Гоше обусловлено мутацией в гене глюкоцереброзидазы (GBA), которая наследуется по аутосомно-рецессивному типу. Мутации гена GBA ведут к недостаточности соответствующего фермента, поэтому процесс разрушения глюкоцереброзидов не осуществляется и гликосфинголипиды накапливаются в макрофагах. Заполненные глюкоцереброзидом макрофаги получили название клеток Гоше. Они обнаруживаются в селезенке, печени, костном мозге, иногда в легких. Этим органам отдается приоритет в ходе клинического обследования при подозрении на болезнь Гоше. Симптомами, которые могут указывать на болезнь Гоше, являются спленомегалия, гепатомегалия, остеонекроз (“инфаркт кости”), тромбоцитопения и/или анемия.
Последний небольшой пример, когда следовало обратить внимание на симптомы лизосомной болезни: у женщины с неотягощенным анамнезом рождается ребенок с несколько завышенными показателями роста и веса (беременность протекала без осложнений). Через три недели мать обратилась к врачу с тем, что ребенок шумно дышит. Позже у ребенка были зафиксированы рецидивирующие случаи бронхита, а в возрасте трех месяцев он поступил в стационар с пневмонией, сопровождаемой обструктивным апноэ. В 11 месяцев ребенку пробовали провести аденотомию, но операция была отложена, так как увеличенный язык мешал доступу; через месяц в другой клинике аденоиды были удалены. Кроме того, бросались в глаза довольно грубые черты лица и большие размеры головы ребенка. На обследовании живот был несколько вздут, имелась небольшая пупочная грыжа. Спленомегалии определено не было, печень увеличена. Также была замечена ограниченность в подвижности суставов и задержка речевого развития. На 21-ом месяце жизни был поставлен диагноз болезнь Хантера (мукополисахаридоз II типа). Диагностически важными симптоматическими показателями данной болезни являются дисморфия, гепатомегалия, контрактуры суставов и отсутствие помутнения роговицы (от других мукополисахаридозов). Результаты лабораторной диагностики указывают на пониженную активность идуронат-2-сульфатазы, а концентрация гликозаминогликанов в моче повышена. Молекулярно-генетическое обследование подтвердит диагноз.
Болезнь Хантера наследуется вместе с Х-хромосомой. Мутация гена идуронатсульфатазы (IDS) ведет к отсутствию фермента – продукта данного гена – или же его сниженной ферментативной активности. Поскольку идуронат-2-сульфатаза в значительной степени задействована в разрушении гликозаминогликанов (GAG), то при ее недостаточности происходит накопление GAG в клетках, что нарушает их деятельность. С повышением концентрации GAG функциональные нарушения органов дыхания, сердечно-сосудистой системы, ЦНС, опорно-двигательной системы нарастают. Прогноз и исход заболевания определяется, в том числе, формой течения, которая может быть нейропатической или не нейропатической. К характерным признакам болезни Хантера можно отнести пупочные или паховые грыжи, тугую подвижность суставов, частые инфекции ЛОР-органов в анамнезе, гепатоспленомегалию, грубые черты лица. Для нейропатической формы заболевания свойственно изначально нормальное развитие ребенка с последующими нарушениями развития, регрессией и деменцией. Энзимотерапия состоит в назначении идурсульфазы, что замедляется прогредиентное течение болезни. И чем раньше терапия была начата, тем более успешны ее результаты.

Таким образом, врачам-педиатрам следует уделять должное внимание, казалось бы, обычным симптомам, а также уметь мыслить системно, собирая из кусочков-симптомов цельные картины истинного диагноза. Критическое мышление и доскональность, проявленные врачом при исследовании, напрямую связаны с тем, какой уровень жизни будет обеспечен его пациенту.
Источник: https://www.aerzteblatt.de/archiv/186475/Seltene-Erkrankungen-(2)-Genau-hinschauen-und-dran-denken